Clear Sky Science · en
Fatty acid oxidation drives acetyl-CoA-dependent H3K9ac reprogramming to promote adaptive resistance to BRAFV600E inhibition in thyroid cancer
Why this matters for people with thyroid cancer
Targeted drugs that home in on a common thyroid cancer mutation, called BRAFV600E, have raised hopes for patients with aggressive disease. Yet many tumors initially shrink and then rebound, finding ways to survive despite treatment. This study uncovers a hidden escape route inside cancer cells: a metabolic detour that rewires how they burn fat and control their genes, ultimately blunting the impact of BRAF-blocking drugs. Understanding this pathway could point to new combination therapies that keep the cancer from bouncing back.
The problem with current targeted treatments
Thyroid cancer is the most frequent cancer of the hormone-producing glands, and while many cases are slow-growing, certain forms can spread quickly and are difficult to treat. In these aggressive tumors, BRAFV600E often acts as a master switch that drives growth, making BRAF-blocking drugs an attractive strategy. However, patients commonly develop resistance: the drugs work at first, then lose effectiveness as surviving cancer cells adapt. Researchers suspected that changes in how the cells handle nutrients and energy might underlie this adaptation, but the details were unclear.
Cancer cells switch fuel to survive
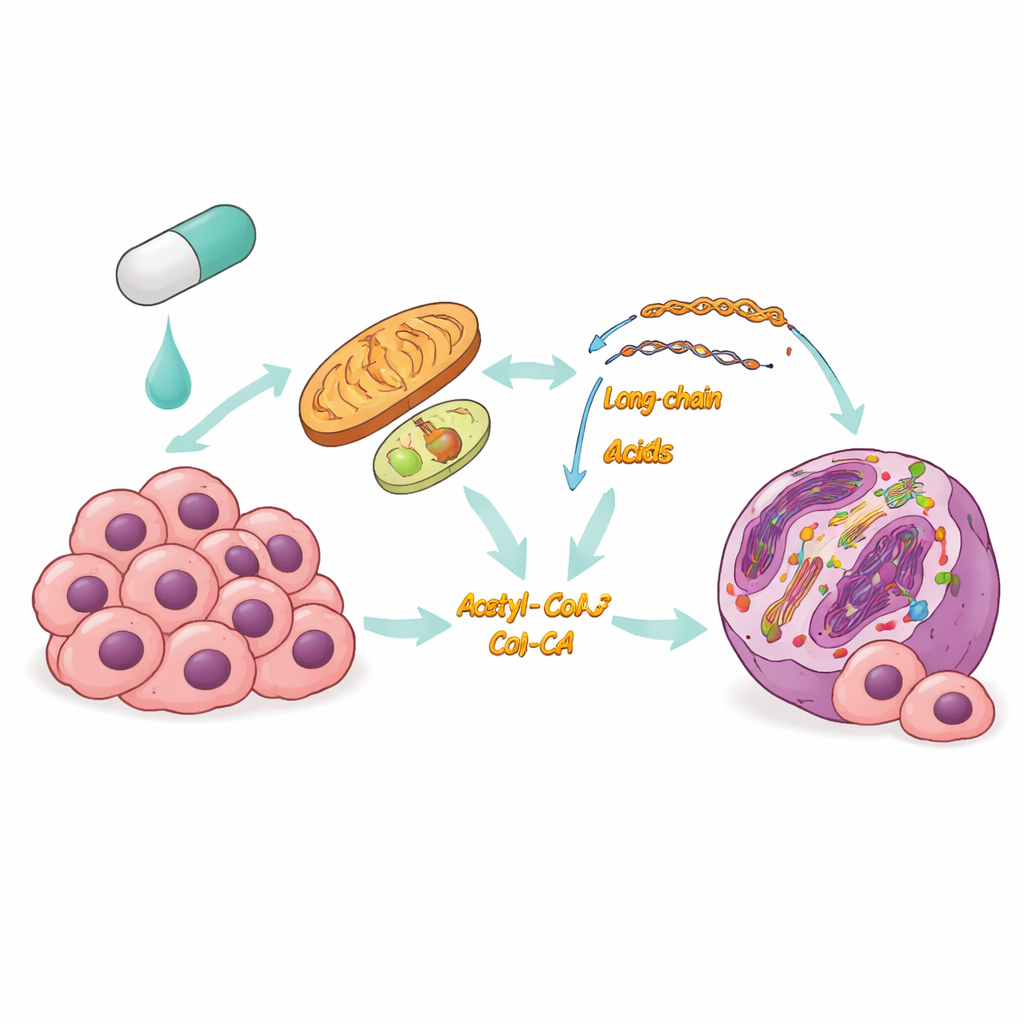
The team used a combination of gene activity profiling and metabolite measurements in thyroid cancer cells exposed to a BRAF inhibitor. They found that, while sugar-burning pathways were dampened, the cells sharply increased fatty acid oxidation—essentially switching from burning glucose to burning fats inside mitochondria and related compartments. Key enzymes that usher fats into this pathway ramped up, lipid droplets were consumed, and both oxygen use and energy output rose. Importantly, this shift was not a fleeting blip: cells that had evolved long-term drug resistance kept fatty acid oxidation turned on, suggesting it is a core survival strategy under BRAF blockade.
A metabolic signal that rewrites the cell’s instruction manual
Burning fats does more than keep the lights on. As fatty acids are broken down, they generate acetyl-CoA, a small molecule that also serves as a building block for chemical tags on DNA-packaging proteins called histones. The researchers showed that elevated fatty acid oxidation under BRAF inhibition boosted acetyl-CoA levels, which in turn increased a specific histone modification known as H3K9 acetylation. This modification loosens local DNA structure and tends to activate nearby genes. When the team blocked fatty acid oxidation with the drug thioridazine, acetyl-CoA fell and this acetylation mark dropped; adding a precursor that replenishes acetyl-CoA partially restored it. Genome-wide mapping revealed that the heightened acetylation clustered near promoters of genes involved in survival and therapy resistance.
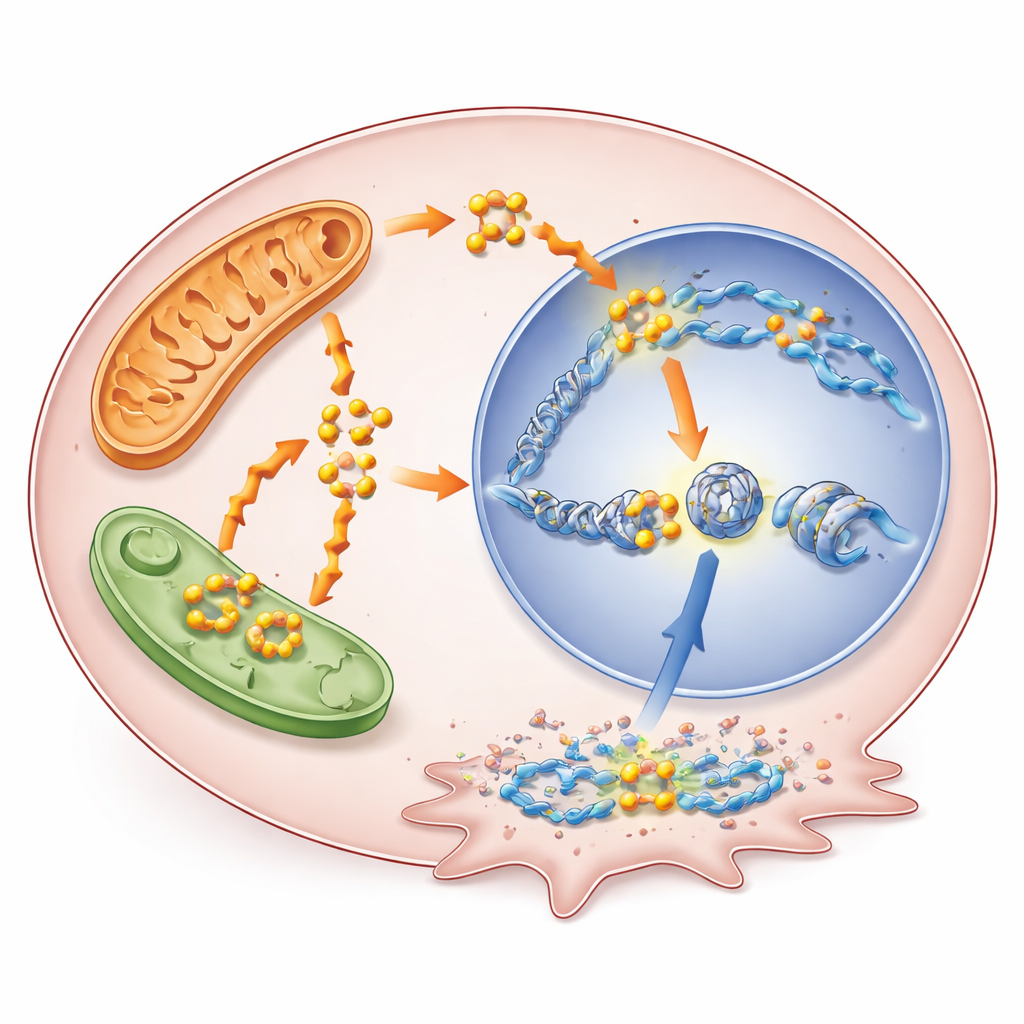
A newly highlighted troublemaker gene
Among the genes switched on in this way, one called RUNX1 stood out. RUNX1 is a transcription factor—a gene-control protein—previously implicated in drug resistance in other cancers, but not well studied in thyroid tumors. Here, its regulatory regions gained extra H3K9 acetylation when BRAF was blocked, and its activity rose in multiple thyroid cancer models, including cells that had become chronically resistant. High RUNX1 levels in patient tumor data correlated with more advanced disease, nodal spread, and poorer outcomes. When the scientists dialed down RUNX1 in thyroid cancer cells, those cells grew more slowly and were less able to migrate and invade, pointing to a direct role in making tumors more aggressive.
Combining treatments to cut off the escape route
Because fatty acid oxidation and RUNX1 both appeared to help cells weather BRAF inhibition, the researchers tested whether blocking these pathways could make existing drugs more potent. In cell lines, pairing a fatty acid oxidation blocker with BRAF inhibitors triggered more cancer cell death than either drug alone. In mice carrying human thyroid tumor grafts, the combination shrank tumors more effectively than BRAF inhibition by itself. A similar strategy boosted the impact of a clinically used doublet of BRAF and MEK inhibitors in a mini-tumor organoid grown from a patient sample. In parallel, a small-molecule inhibitor of RUNX1 made thyroid cancer cells more vulnerable to BRAF-targeted therapy, reinforcing the idea that this metabolic–epigenetic axis is therapeutically exploitable.
What this means for future care
For non-specialists, the key message is that some thyroid cancers survive targeted treatment by changing both their fuel choice and how their DNA is read. By burning more fat, the cells generate a chemical signal that opens up survival-promoting genes like RUNX1, helping them adapt to BRAF-blocking drugs. Interrupting this chain—either by blocking fatty acid breakdown or by dampening RUNX1’s activity—makes the cancer cells more likely to die under treatment. While these findings are still preclinical, they outline a clear rationale for future trials that combine metabolism-focused drugs with existing targeted therapies to delay or overcome resistance in aggressive thyroid cancer.
Citation: Wang, X., Zhang, J., Yuan, J. et al. Fatty acid oxidation drives acetyl-CoA-dependent H3K9ac reprogramming to promote adaptive resistance to BRAFV600E inhibition in thyroid cancer. Cell Death Dis 17, 329 (2026). https://doi.org/10.1038/s41419-026-08575-7
Keywords: thyroid cancer, BRAF inhibition, fatty acid oxidation, epigenetic reprogramming, drug resistance