Clear Sky Science · en
BMX inhibition overcomes small cell lung cancer chemoresistance by stabilizing E2F1 via ERK1/2-Cyclin D1/CDK4/6 axis
Why this matters for people with lung cancer
Small cell lung cancer is one of the deadliest forms of lung cancer. It often shrinks at first when hit with chemotherapy, only to come roaring back in a form that no longer responds to treatment. This study uncovers a key molecular “escape route” that tumor cells use to survive chemotherapy and introduces a new drug candidate designed to block that route. By showing how to re-sensitize stubborn tumors to standard drugs, the work points toward more durable treatment options for patients.
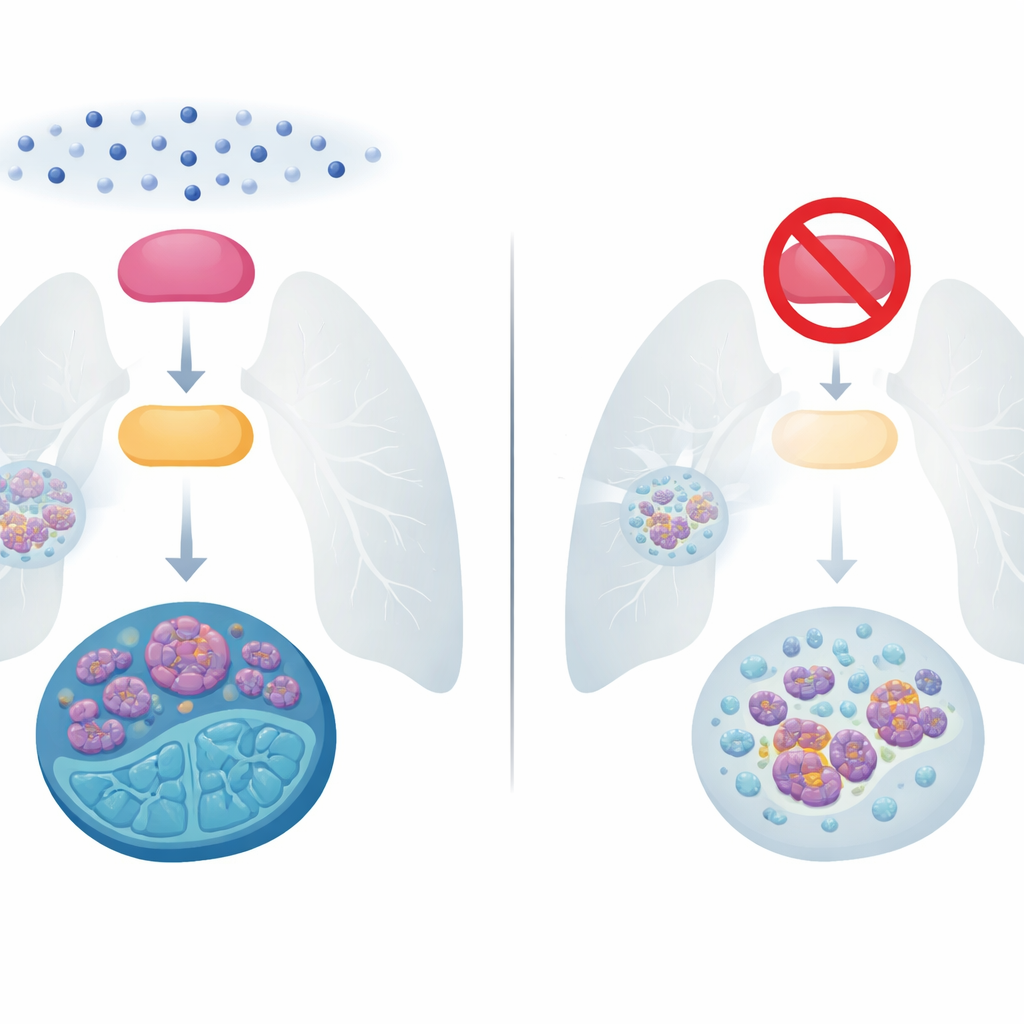
The challenge of runaway small cell lung cancer
Small cell lung cancer accounts for about 15% of lung cancer cases but causes a disproportionate number of deaths because it grows and spreads quickly. For decades, the main treatment has been a combination of platinum-based chemotherapy and the drug etoposide. Many patients initially respond well, but most tumors soon return in a harder-to-treat form, and five-year survival remains under 10%. Newer approaches, including immunotherapy, have only modestly extended life for people with advanced disease. Understanding exactly how these tumors become resistant to chemotherapy is therefore one of the most urgent questions in this field.
A survival switch inside tumor cells
The researchers focused on two proteins that act like a signaling switchboard inside cancer cells. One, called BMX, is a kinase—a kind of enzyme that flips other proteins on or off. The other, E2F1, is a transcription factor that turns on genes controlling cell division, DNA repair, and the ability of cells to move and invade. In samples from 100 patients and in multiple small cell lung cancer cell lines, the team found that active BMX and high levels of E2F1 almost always went together, especially in tumors that had stopped responding to chemotherapy. When cells were exposed to common drugs such as cisplatin or etoposide, both BMX activity and E2F1 levels rose in lockstep, and drug‑resistant cell lines showed much higher levels of both proteins than their drug‑sensitive counterparts.
How the tumor’s inner wiring protects it from drugs
Diving deeper, the team uncovered how BMX helps E2F1 stay active. Rather than binding E2F1 directly, BMX turns on a relay of other proteins—ERK1/2 and the Cyclin D1/CDK4/6 complex—that chemically modify E2F1 at specific positions. These modifications act like protective armor, making E2F1 more stable and helping it accumulate in the cell nucleus, where it can switch on genes for rapid growth, efficient DNA repair, and cell migration. Normally, E2F1 can be tagged for destruction by the cell’s protein‑recycling machinery. In chemoresistant cells, however, BMX signaling shields E2F1 from this disposal system, so the protein persists and helps tumor cells survive chemotherapy‑induced damage.
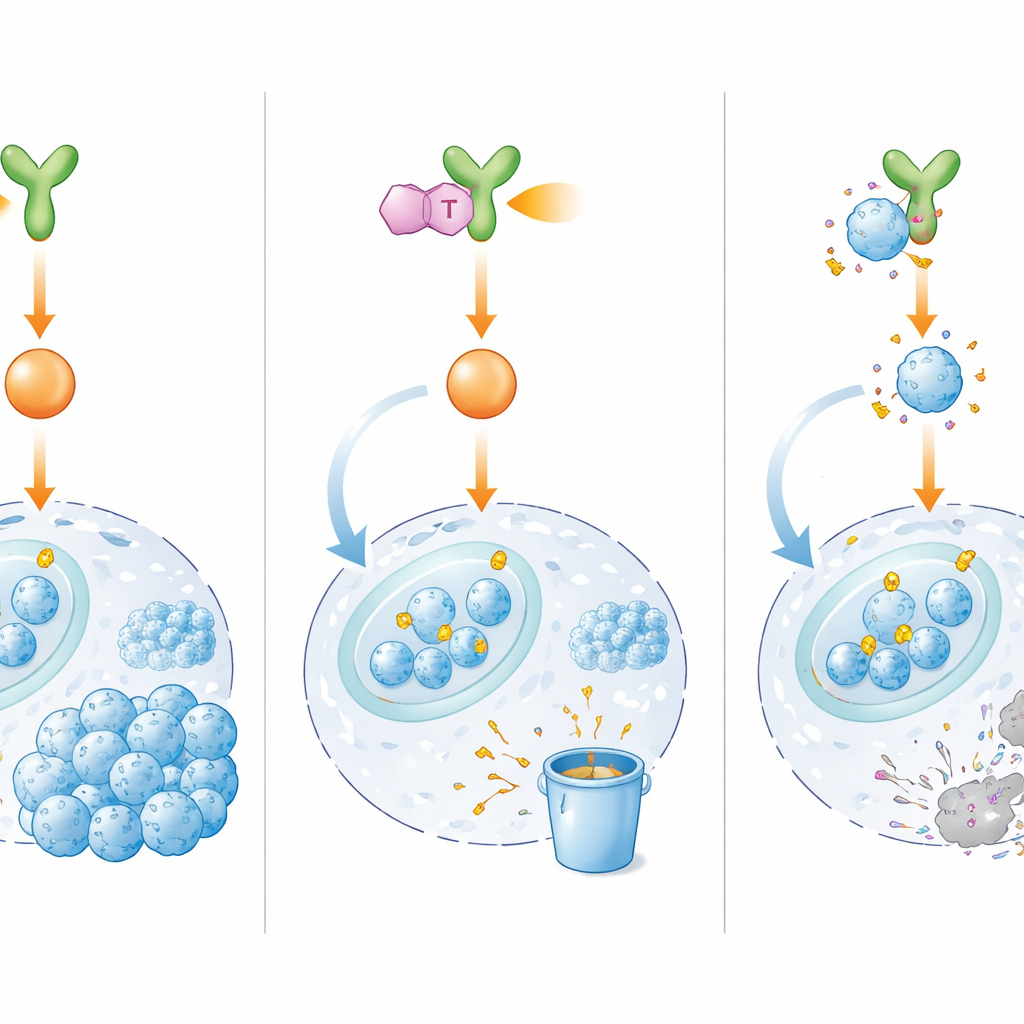
A new drug that cuts the power to the switch
Because directly targeting E2F1 has proven difficult, the researchers set out to block its upstream controller, BMX. From thousands of candidate molecules, they identified IHMT‑15137, a small compound that binds strongly and selectively to BMX and appears to latch onto it irreversibly. In laboratory tests, IHMT‑15137 shut down BMX activity and dimmed its downstream signals, including ERK1/2 and Cyclin D1/CDK4/6. As a result, E2F1 lost its protective chemical marks, became more heavily tagged for destruction, and was rapidly broken down. In multiple drug‑resistant cell lines, as well as in patient‑derived cells and three‑dimensional mini‑tumors, combining IHMT‑15137 with cisplatin led to stronger cell‑cycle arrest, more DNA damage, higher rates of cell death, and sharply reduced growth, migration, and invasion compared with chemotherapy alone.
Proof of benefit in animal and patient‑like models
The team then tested the combination in mice carrying human small cell lung cancer grafts, including tumors directly derived from patients whose disease had relapsed after chemotherapy. In several independent models, adding IHMT‑15137 to standard cisplatin–etoposide therapy significantly slowed tumor growth without causing substantial weight loss or clear organ damage in the animals. Tumors from treated mice showed reduced activation of BMX and its signaling partners, lower E2F1 levels, and more markers of DNA damage and apoptosis. Safety studies suggested that IHMT‑15137 has fewer bleeding and off‑target effects than older, less selective drugs that also hit BMX, supporting its potential as a more tolerable partner for chemotherapy.
What this means for future treatment
To a non‑specialist, the key message is that the researchers have traced a major resistance pathway in small cell lung cancer and shown how to shut it down. Tumor cells use BMX to stabilize E2F1, a master growth regulator, helping them repair chemotherapy‑induced damage and keep dividing. The new inhibitor IHMT‑15137 cuts power to this pathway, destabilizing E2F1 and making long‑resistant tumors vulnerable again to existing drugs. While this compound will need further optimization and human trials, the study provides a clear blueprint for therapies that could extend and deepen chemotherapy responses for patients with this aggressive cancer.
Citation: Wu, T., Qi, S., Shi, C. et al. BMX inhibition overcomes small cell lung cancer chemoresistance by stabilizing E2F1 via ERK1/2-Cyclin D1/CDK4/6 axis. Sig Transduct Target Ther 11, 125 (2026). https://doi.org/10.1038/s41392-026-02644-1
Keywords: small cell lung cancer, chemoresistance, BMX kinase, E2F1 signaling, targeted therapy