Clear Sky Science · zh
化学空间边缘的分子深度学习
为何更聪明的药物预测很重要
现代药物发现越来越依赖人工智能来筛选庞大的化合物库并标出有前景的候选药物。但有一个问题:许多机器学习模型只在与训练时见过的分子非常相似的情况下表现良好。当被要求评估更不寻常的化合物——恰恰可能成为首创药物的那些——这些模型可能会过于自信并给出错误判断。本研究提出了一种判定模型何时处于不稳固状态的新方法,帮助研究人员安全地推进到化学空间尚未探明的区域。
当地图不再可用
在早期药物发现中,科学家寻找“命中(hits)”:能够影响与疾病相关的蛋白等生物靶点的小分子。由于在实验室上测试数十亿种可能的分子不现实,机器学习模型通常在几百或几千种已知化合物上训练,然后用于预测哪些新化合物值得实验验证。然而,当遇到与训练数据不同的分子时,这些模型往往会失效——这被称为分布转移。现有的安全措施要么在已知区域周围划出一条硬边界,从而阻止更冒险的化合物,要么以可能在模型遇到真正新颖样本时仍然误导人的方式来估计预测不确定性。
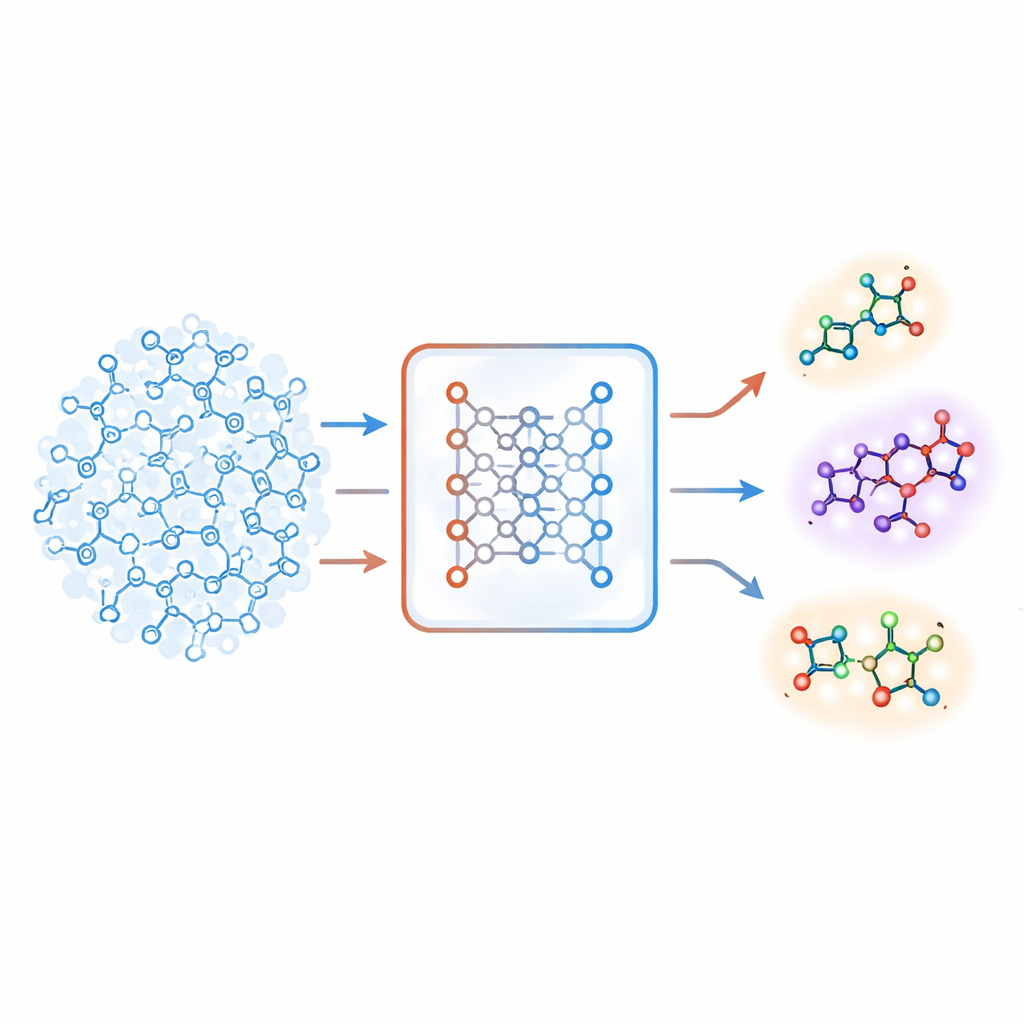
一种新的不熟悉感
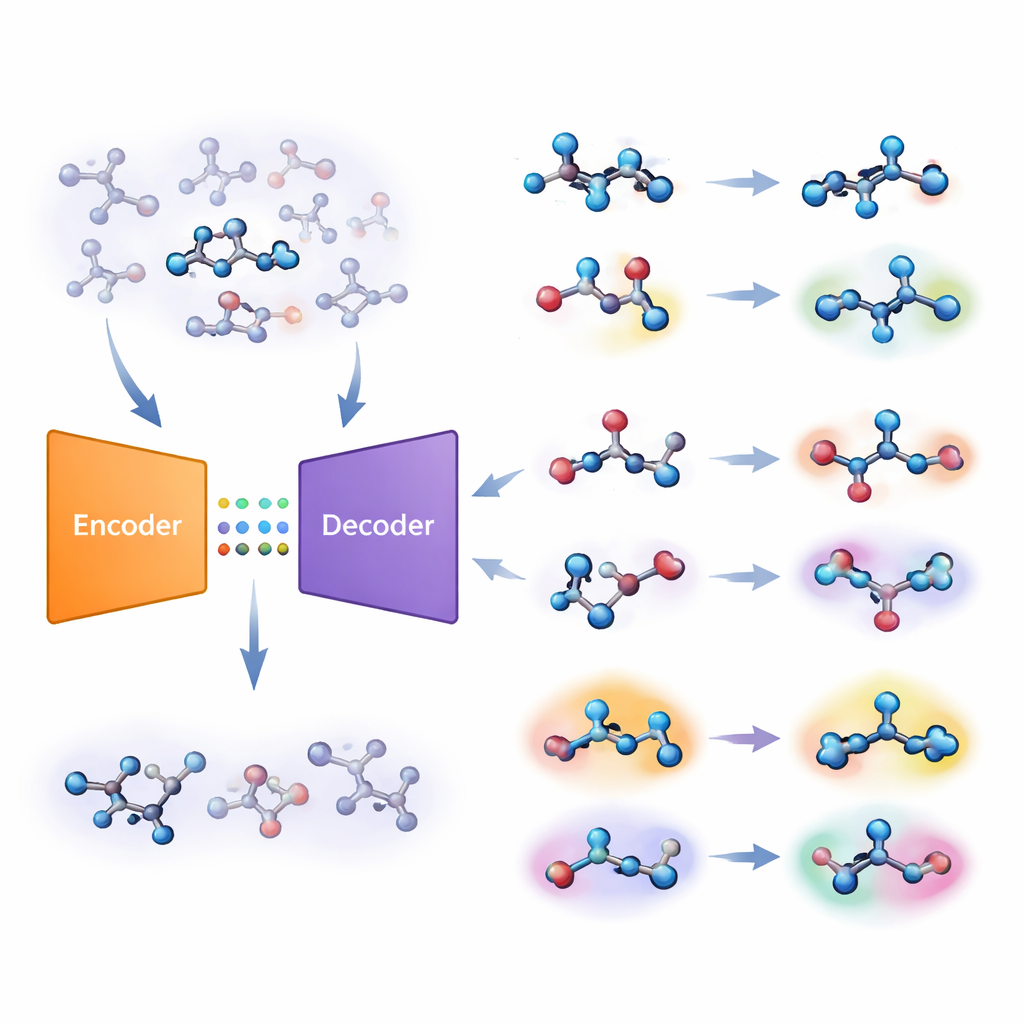
作者提出了一种基于自编码器(一类深度学习系统)的不同策略。他们的“联合分子模型”同时学习两项任务:预测某分子在靶点上的活性,以及从压缩的内部编码重建该分子。如果模型难以准确重建某个分子,则该分子被视为“不熟悉”。团队将这种重建误差转化为一个数值分数,称为不熟悉度,用以反映一个分子距模型实际学到的化学模式有多远。关键在于,这个分数由模型自身对化学的理解驱动,而不是简单的手工设计的相似性度量。
测试化学空间的边界
为了检验不熟悉度在检测模型能力边界时的表现,研究人员收集了涵盖不同生物靶点和性质的33个数据集。他们使用聚类方法将每个数据集划分为典型样本和结构上更不寻常的样本,模拟了已充分研究分子与新奇分子之间的差异。在这些基准测试中,被标记为分布外的分子一致表现出更高的不熟悉度。这一效应不能仅用分子大小或复杂度等琐碎特征来解释。相反,不熟悉度与分子的结构核心距训练化合物的距离密切相关,证实了模型能够有效感知分子有多“超出图表”。
发现仅靠不确定性可能错过的东西
研究团队随后将不熟悉度与几种常见的预测可靠性评估方法进行了比较,包括模型不确定性和各种化学相似性度量。无论是不熟悉度还是不确定性,都与分类器的性能有关:当任一指标较高时,预测往往更不准确。然而,这两种信号在很大程度上是独立的。不熟悉度同时捕捉到了结构距离和性能信息,而单独的不确定性并不能很好地反映结构,尤其是在分子来自截然不同分布时。在对超过一百万种商业分子的巨大虚拟筛选中,不熟悉度能明显区分常规化合物与真正新颖的化合物,而不确定性则几乎未能区分这两类。
从屏幕到湿实验室
为展示实际影响,研究人员对大约18万种可购分子进行了前瞻性筛选,寻找两种与疾病相关酶(PIM1 和 CDK1)的抑制剂。他们在适度现有数据集上训练联合模型,然后同时用三项信息对新化合物排序:预测活性、模型不确定性和不熟悉度。在仅购买并在生化测定中测试60种分子后,他们发现了七种低微摩尔效力的化合物,且这些化合物在结构上与训练集化合物和典型的激酶抑制剂明显不同。那些倾向于低不熟悉度(但仍允许一定不确定性)的策略往往产生更强的命中,表明关注不熟悉度可以引导探索走向有前景而又不完全陌生的化学区域。
对未来药物的意义
通俗地说,不熟悉度评分为化学领域的机器学习模型提供了一种内置的感知,告诉它们何时正在从已知范围外进行外推。通过将这种感知与模型重建分子的能力相联系,这种方法同时反映了化学相似性和预测可信度。研究表明,这一指标能发现标准方法漏检的分布转移,改善虚拟筛选中的优先级排序,并在实际实验中帮助发现新的化学物质。随着药物研发者越来越多地进军广阔且大部分未被绘制的化学空间,不熟悉度为决定哪些大胆预测值得相信并送入实验室验证提供了一个有原则的指南针。
引用: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
关键词: 分子机器学习, 药物发现, 化学空间, 分布外, 虚拟筛选