Clear Sky Science · es
Aprendizaje profundo molecular en el límite del espacio químico
Por qué importan predicciones de fármacos más inteligentes
El descubrimiento moderno de fármacos recurre cada vez más a la inteligencia artificial para rastrear enormes bibliotecas de compuestos y señalar candidatos prometedores. Pero hay un problema: muchos modelos de aprendizaje automático funcionan bien solo con moléculas que se parecen mucho a las que han visto antes. Cuando se les pide evaluar compuestos más inusuales —justo aquellos que podrían convertirse en medicinas primeras en su clase— estos modelos pueden mostrarse excesivamente confiados y equivocarse. Este estudio presenta una nueva forma de saber cuándo un modelo está en terreno inestable, ayudando a los investigadores a avanzar con seguridad hacia regiones inexploradas del espacio químico.
Cuando el mapa se agota
En la etapa temprana del descubrimiento de fármacos, los científicos buscan “hits”: pequeñas moléculas que afectan a un objetivo biológico como una proteína relacionada con una enfermedad. Dado que realizar experimentos de laboratorio con miles de millones de moléculas posibles es imposible, se entrenan modelos de aprendizaje automático con unos pocos cientos o miles de compuestos conocidos y luego se usan para predecir cuáles merecen pruebas adicionales. Sin embargo, estos modelos tienden a fallar con moléculas distintas a las de sus datos de entrenamiento, un problema conocido como cambio de distribución. Las salvaguardas existentes o bien dibujan un límite rígido alrededor de la región conocida, bloqueando moléculas más aventureras, o estiman la incertidumbre de la predicción de maneras que pueden seguir siendo engañosas cuando el modelo encuentra algo verdaderamente nuevo.
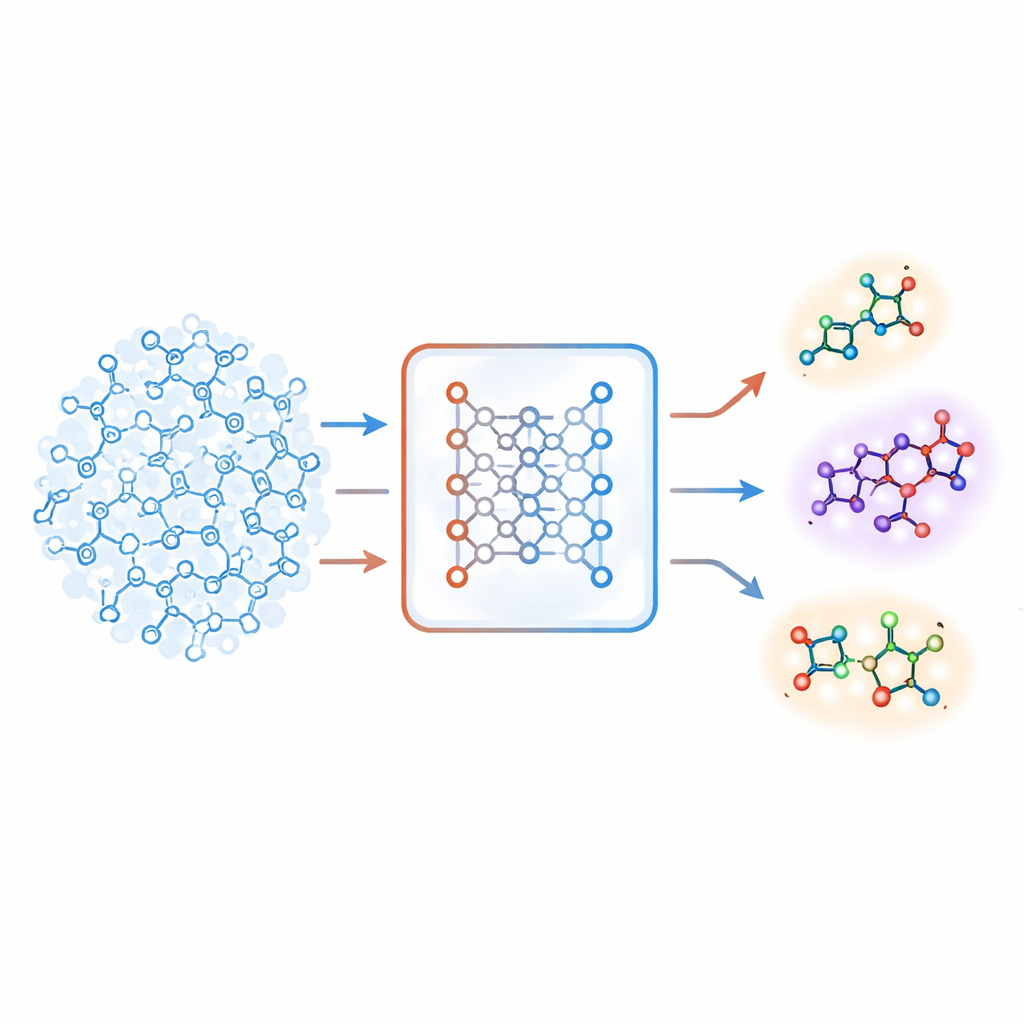
Un nuevo sentido de lo desconocido
Los autores proponen una estrategia distinta basada en un tipo de sistema de aprendizaje profundo llamado autoencoder. Su “modelo molecular conjunto” aprende dos tareas a la vez: predecir si una molécula es activa sobre un objetivo y reconstruir la molécula a partir de un código interno comprimido. Si el modelo tiene dificultades para reconstruir con precisión una molécula concreta, esa molécula se considera “desconocida”. El equipo convierte este error de reconstrucción en una puntuación numérica, llamada unfamiliarity (desconocimiento), que refleja qué tan alejada está una molécula de los patrones químicos que el modelo ha aprendido. De forma crucial, esta puntuación se basa en la propia comprensión química del modelo y no en medidas de similitud simples y diseñadas a mano.
Probar el límite del espacio químico
Para sondear qué tan bien el desconocimiento detecta cuando un modelo está fuera de su profundidad, los investigadores reunieron 33 conjuntos de datos que cubrían distintos objetivos biológicos y propiedades. Usaron métodos de agrupamiento para dividir cada conjunto en ejemplos típicos y en otros más estructuralmente inusuales, imitando la diferencia entre moléculas bien estudiadas y novedosas. En estos puntos de referencia, las moléculas etiquetadas como fuera de distribución presentaron sistemáticamente puntuaciones de desconocimiento más altas. Este efecto no pudo explicarse por características triviales como el tamaño o la complejidad molecular. En cambio, el desconocimiento siguió de cerca la distancia entre el núcleo estructural de una molécula y el de los compuestos de entrenamiento, confirmando que el modelo captaba efectivamente qué tan “fuera de la gráfica” estaba una molécula.
Ver lo que la incertidumbre por sí sola puede pasar por alto
El equipo comparó entonces el desconocimiento con varias formas comunes de juzgar la fiabilidad de una predicción, incluida la incertidumbre del modelo y diversas medidas de similitud química. Tanto el desconocimiento como la incertidumbre se relacionaron con el rendimiento del clasificador: cuando cualquiera de las métricas era alta, las predicciones tendían a ser menos precisas. Sin embargo, las dos señales eran en gran medida independientes. El desconocimiento capturó tanto la distancia estructural como el rendimiento, mientras que la incertidumbre por sí sola no reflejaba bien la estructura, especialmente cuando las moléculas procedían de una distribución muy diferente. En cribados virtuales masivos de más de un millón de moléculas comerciales, el desconocimiento separó claramente compuestos rutinarios de los genuinamente novedosos, mientras que la incertidumbre sugería poca diferencia entre ambos grupos.
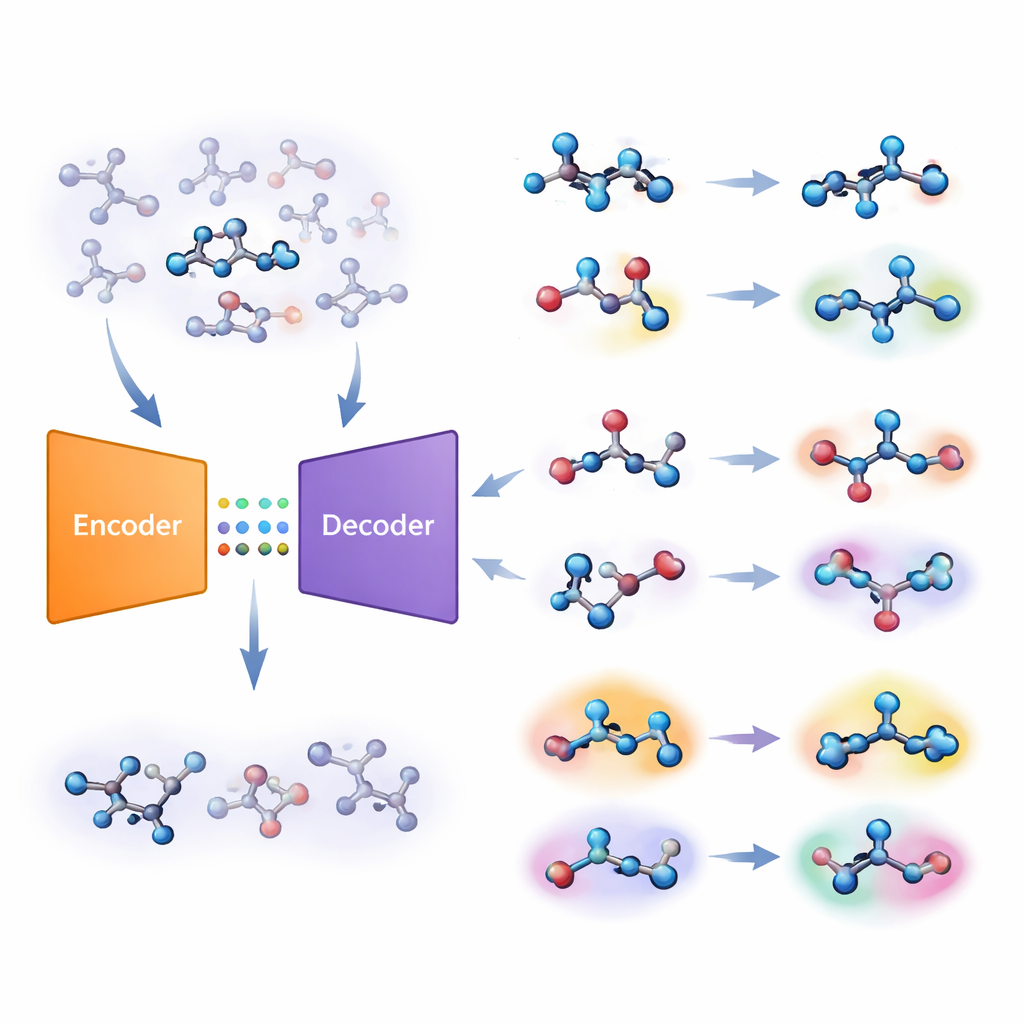
De la pantalla del ordenador al laboratorio experimental
Para demostrar el impacto práctico, los investigadores realizaron un cribado prospectivo sobre unas 180.000 moléculas adquiribles, buscando inhibidores de dos enzimas relevantes para la enfermedad, PIM1 y CDK1. Entrenaron su modelo conjunto con conjuntos de datos existentes modestos y luego clasificaron nuevos compuestos usando tres ingredientes a la vez: actividad predicha, incertidumbre del modelo y desconocimiento. Tras comprar y probar solo 60 moléculas en ensayos bioquímicos, descubrieron siete con potencia en el rango micromolar bajo, todas estructuralmente distintas de los compuestos de entrenamiento y de los inhibidores típicos de quinasas. Las estrategias que priorizaron bajo desconocimiento —sin excluir la incertidumbre— tendieron a producir los hits más potentes, lo que sugiere que atender al desconocimiento puede guiar la exploración hacia química prometedora pero no completamente ajena.
Lo que esto significa para futuros medicamentos
En términos cotidianos, la puntuación de desconocimiento da a los modelos de aprendizaje automático para química una sensación integrada de cuándo están extrapolando demasiado lejos de lo que saben. Al ligar esta sensación a la capacidad del modelo para reconstruir moléculas, el enfoque refleja simultáneamente la similitud química y la confiabilidad de la predicción. El estudio muestra que esta métrica puede revelar cambios de distribución que los métodos estándar pasan por alto, mejorar la priorización en cribados virtuales y ayudar a descubrir materia química fresca en experimentos reales. A medida que los cazadores de fármacos se adentran cada vez más en las vastas y en gran parte inexploradas regiones del espacio químico, el desconocimiento ofrece una brújula fundamentada para decidir qué predicciones audaces merecen crédito y ser probadas en el laboratorio.
Cita: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
Palabras clave: aprendizaje automático molecular, descubrimiento de fármacos, espacio químico, fuera de distribución, cribado virtual