Clear Sky Science · en
Molecular deep learning at the edge of chemical space
Why smarter drug predictions matter
Modern drug discovery increasingly leans on artificial intelligence to sift through enormous libraries of chemicals and flag promising drug candidates. But there is a catch: many machine-learning models only work well on molecules that look very similar to those they have seen before. When asked to judge more unusual compounds—the very ones that might become first-in-class medicines—these models can become overconfident and wrong. This study introduces a new way to tell when a model is on shaky ground, helping researchers push safely into unexplored regions of chemical space.
When the map runs out
In early-stage drug discovery, scientists search for “hits”: small molecules that affect a biological target such as a protein linked to disease. Because running lab experiments on billions of possible molecules is impossible, machine-learning models are trained on a few hundred or thousand known compounds and then used to predict which new ones are worth testing. However, these models tend to break down on molecules unlike those in their training data—a problem known as distribution shift. Existing safeguards either draw a hard boundary around the known region, blocking more adventurous molecules, or estimate prediction uncertainty in ways that can still be misleading when the model sees something truly new.
A new sense of unfamiliarity
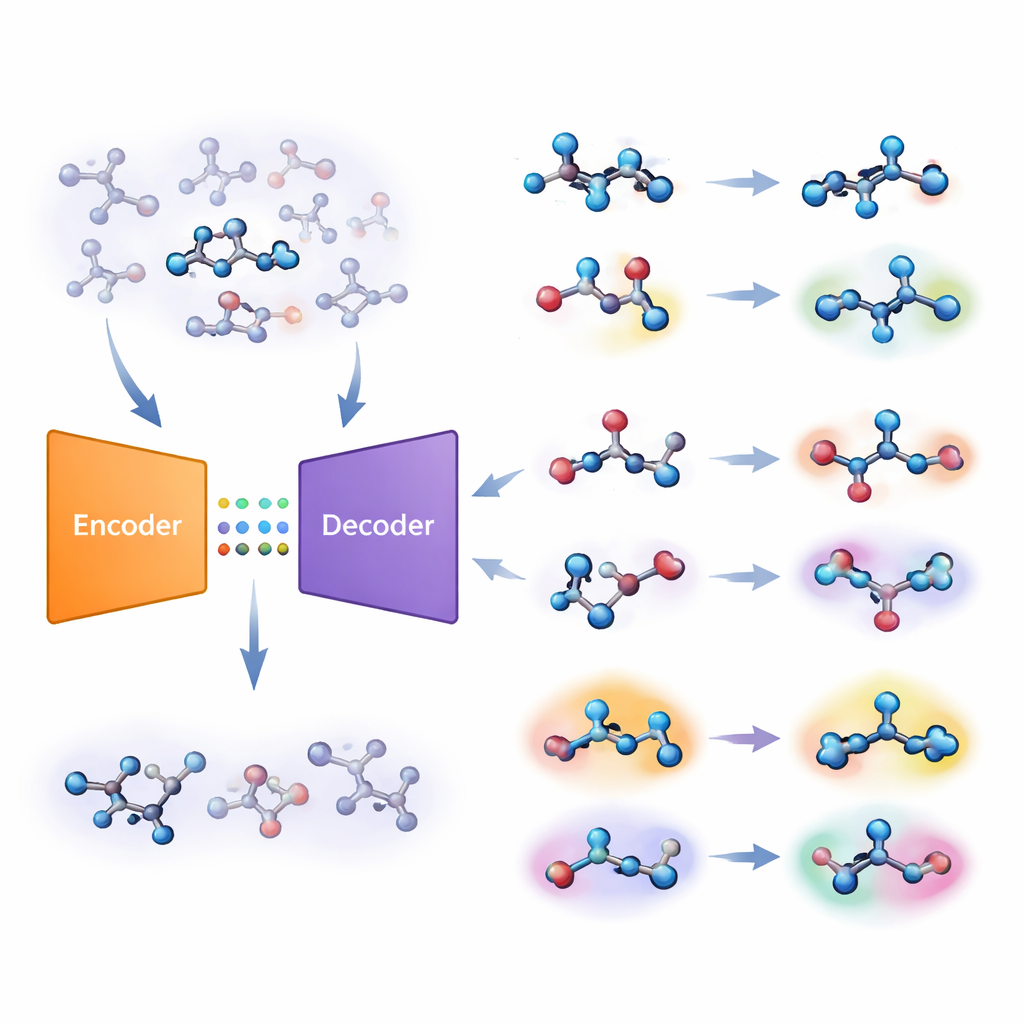
The authors propose a different strategy built on a type of deep-learning system called an autoencoder. Their “joint molecular model” learns two tasks at once: predicting whether a molecule is active on a target, and reconstructing the molecule from a compressed internal code. If the model struggles to rebuild a particular molecule accurately, that molecule is deemed “unfamiliar.” The team turns this reconstruction error into a numerical score, called unfamiliarity, that reflects how far a molecule lies from the chemical patterns the model has actually learned. Crucially, this score is driven by the model’s own understanding of chemistry rather than by simple, hand-crafted similarity measures.
Testing the edge of chemical space
To probe how well unfamiliarity detects when a model is out of its depth, the researchers assembled 33 datasets covering different biological targets and properties. They used clustering methods to split each dataset into typical examples and more structurally unusual ones, mimicking the difference between well-studied and novel molecules. Across these benchmarks, molecules tagged as out-of-distribution consistently had higher unfamiliarity scores. This effect could not be explained away by trivial features such as molecule size or complexity. Instead, unfamiliarity closely tracked how far a molecule’s structural core lay from that of the training compounds, confirming that the model was effectively sensing how “off the chart” a molecule was.
Seeing what uncertainty alone can miss
The team then compared unfamiliarity with several common ways of judging prediction reliability, including model uncertainty and various measures of chemical similarity. Both unfamiliarity and uncertainty were linked to how well the classifier performed: when either metric was high, predictions tended to be less accurate. Yet the two signals were largely independent. Unfamiliarity captured both structural distance and performance, while uncertainty alone did not reflect structure very well, especially when molecules came from a very different distribution. In massive virtual screens of more than a million commercial molecules, unfamiliarity sharply separated routine compounds from genuinely novel ones, whereas uncertainty suggested little difference between the two groups.
From computer screen to wet lab
To demonstrate practical impact, the researchers ran a prospective screen on about 180,000 purchasable molecules, searching for inhibitors of two disease-relevant enzymes, PIM1 and CDK1. They trained their joint model on modest existing datasets and then ranked new compounds using three ingredients at once: predicted activity, model uncertainty and unfamiliarity. After buying and testing only 60 molecules in biochemical assays, they discovered seven with low micromolar potency, all structurally distinct from the training compounds and typical kinase inhibitors. Strategies that favored low unfamiliarity—while still allowing uncertainty—tended to produce the strongest hits, suggesting that paying attention to unfamiliarity can guide exploration toward promising yet not completely alien chemistry.
What this means for future medicines
In everyday terms, the unfamiliarity score gives machine-learning models for chemistry a built-in sense of when they are extrapolating too far from what they know. By tying this sense to the model’s ability to reconstruct molecules, the approach simultaneously reflects chemical similarity and prediction trustworthiness. The study shows that this metric can uncover distribution shifts that standard methods miss, improve prioritization in virtual screens and help uncover fresh chemical matter in real experiments. As drug hunters increasingly venture into the vast, largely uncharted reaches of chemical space, unfamiliarity offers a principled compass for deciding which bold predictions are worth believing—and testing in the lab.
Citation: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
Keywords: molecular machine learning, drug discovery, chemical space, out-of-distribution, virtual screening