Clear Sky Science · it
Apprendimento profondo molecolare al confine dello spazio chimico
Perché contano previsioni farmacologiche più intelligenti
La scoperta di farmaci moderna si affida sempre più all’intelligenza artificiale per setacciare enormi librerie di sostanze chimiche e segnalare candidati promettenti. C’è però un problema: molti modelli di machine learning funzionano bene solo su molecole che somigliano molto a quelle viste in fase di addestramento. Quando devono valutare composti più insoliti — proprio quelli che potrebbero diventare farmaci first-in-class — questi modelli possono mostrarsi eccessivamente sicuri e sbagliare. Questo studio introduce un nuovo modo per capire quando un modello si trova su terreno instabile, aiutando i ricercatori a spingersi in modo sicuro in regioni inesplorate dello spazio chimico.
Quando la mappa finisce
Nelle fasi iniziali della scoperta di farmaci, gli scienziati cercano “hit”: piccole molecole che influenzano un bersaglio biologico come una proteina legata a una malattia. Poiché è impossibile eseguire esperimenti di laboratorio su miliardi di possibili molecole, i modelli di machine learning vengono addestrati su poche centinaia o migliaia di composti noti e poi usati per prevedere quali nuovi composti vale la pena testare. Tuttavia, questi modelli tendono a degradare le prestazioni su molecole diverse da quelle presenti nei dati di addestramento — un problema noto come shift di distribuzione. Le salvaguardie esistenti o tracciano un confine netto intorno alla regione nota, bloccando molecole più avventurose, o stimano l’incertezza delle previsioni in modi che possono comunque risultare fuorvianti quando il modello incontra qualcosa di veramente nuovo.
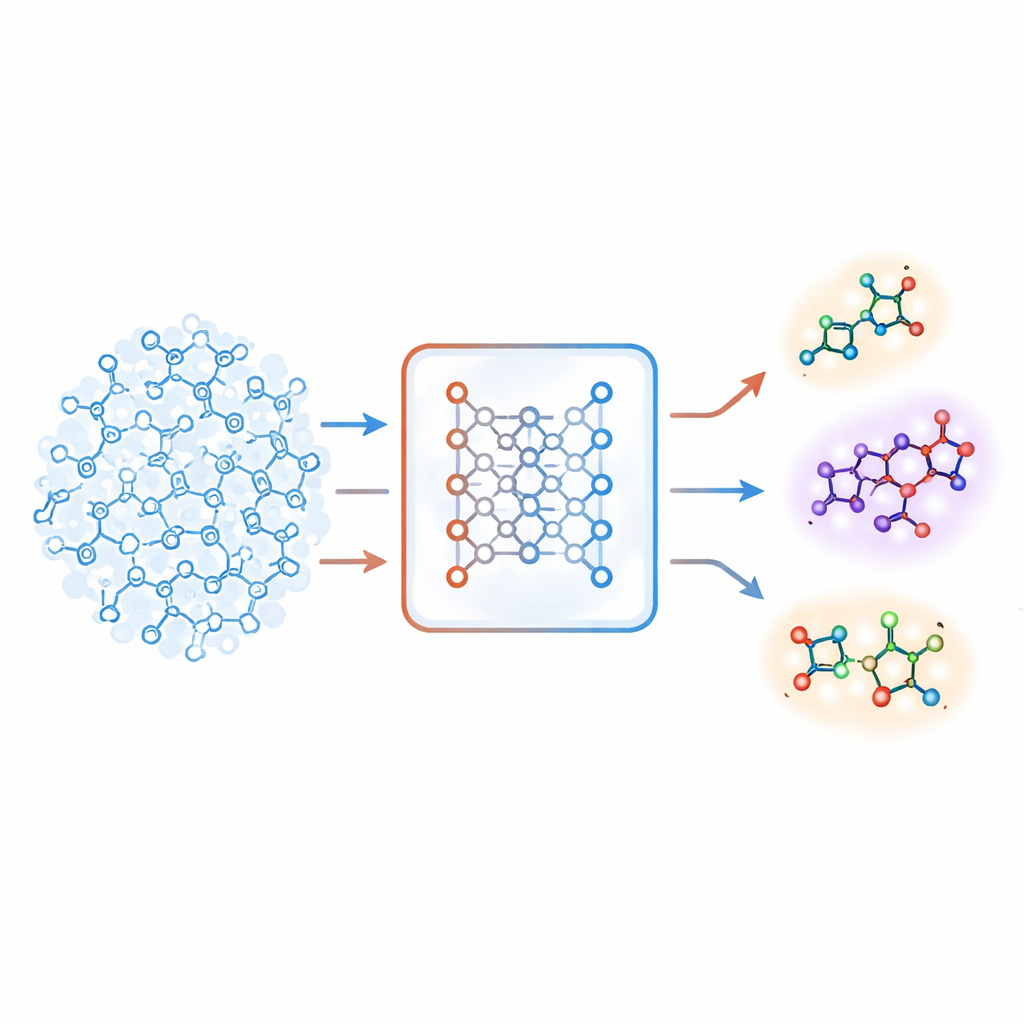
Un nuovo senso dell’estraneità
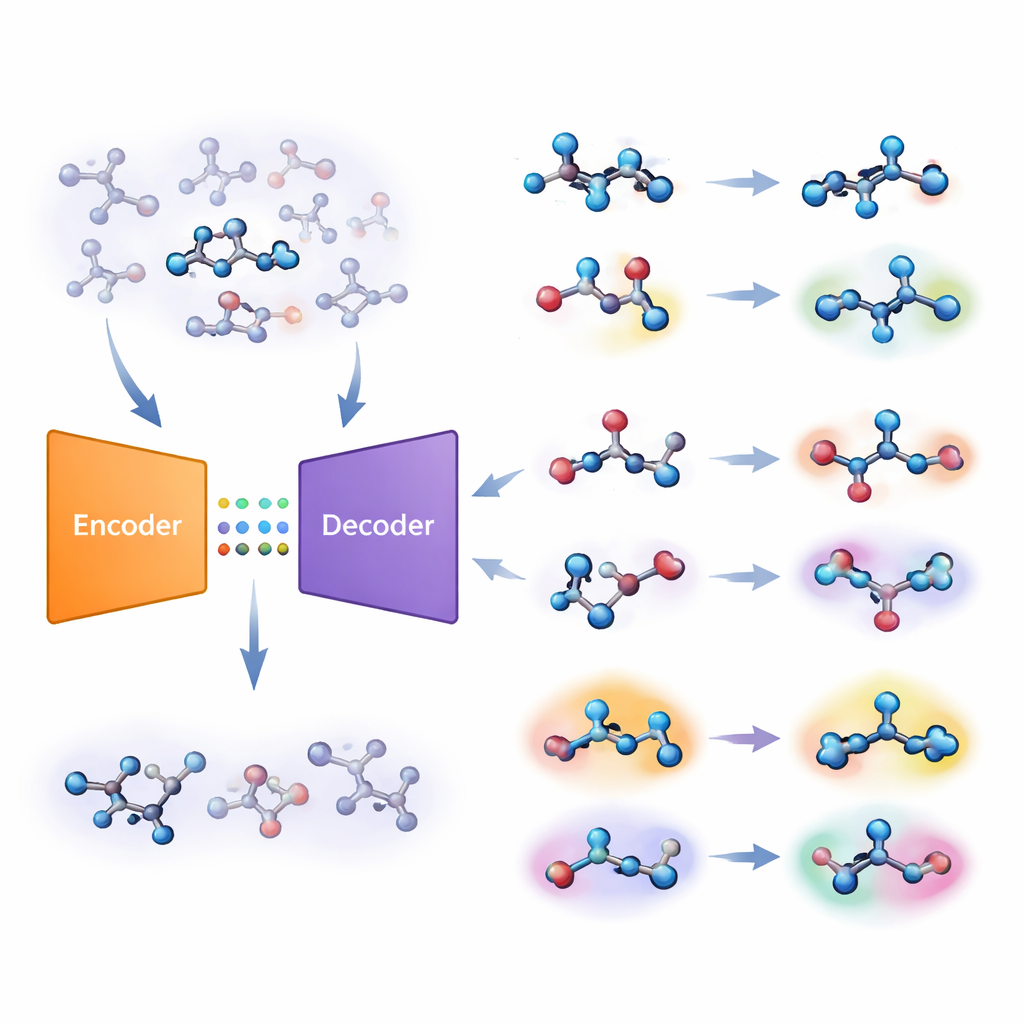
Gli autori propongono una strategia diversa basata su un tipo di sistema di deep learning chiamato autoencoder. Il loro “modello molecolare congiunto” apprende due compiti contemporaneamente: predire se una molecola è attiva su un bersaglio e ricostruire la molecola a partire da un codice interno compresso. Se il modello fatica a ricostruire accuratamente una certa molecola, quella molecola viene considerata “estranea”. Il team trasforma questo errore di ricostruzione in un punteggio numerico, chiamato estraneità, che riflette quanto una molecola si discosta dai pattern chimici che il modello ha effettivamente appreso. Fondamentale è che questo punteggio sia guidato dalla comprensione chimica interna del modello piuttosto che da semplici misure di similarità costruite a mano.
Testare il confine dello spazio chimico
Per valutare quanto bene l’estraneità rilevi quando un modello è fuori dal suo elemento, i ricercatori hanno assemblato 33 dataset che coprono diversi bersagli biologici e proprietà. Hanno usato metodi di clustering per dividere ogni dataset in esempi tipici e in altri più strutturalmente insoliti, imitando la differenza tra molecole ben studiate e molecole nuove. In questi benchmark, le molecole etichettate come fuori-distribuzione avevano consistentemente punteggi di estraneità più alti. Questo effetto non poteva essere spiegato da caratteristiche banali come la dimensione o la complessità della molecola. Invece, l’estraneità seguiva da vicino quanto il nucleo strutturale di una molecola fosse distante da quello dei composti di addestramento, confermando che il modello percepiva efficacemente quanto una molecola fosse “fuori dalla mappa”.
Ciò che l’incertezza da sola può perdere
Il team ha quindi confrontato l’estraneità con diversi metodi comuni per valutare l’affidabilità delle previsioni, inclusa l’incertezza del modello e varie misure di similarità chimica. Sia l’estraneità sia l’incertezza erano correlati alle prestazioni del classificatore: quando uno dei due indicatori era alto, le previsioni tendevano a essere meno accurate. Tuttavia i due segnali erano in gran parte indipendenti. L’estraneità catturava sia la distanza strutturale sia le prestazioni, mentre l’incertezza da sola non rifletteva molto bene la struttura, specialmente quando le molecole provenivano da una distribuzione molto diversa. In grandi screening virtuali di oltre un milione di molecole commerciali, l’estraneità separava nettamente i composti di routine da quelli genuinamente nuovi, mentre l’incertezza suggeriva poca differenza fra i due gruppi.
Dallo schermo del computer al laboratorio
Per dimostrare l’impatto pratico, i ricercatori hanno condotto uno screening prospettico su circa 180.000 molecole acquistabili, cercando inibitori di due enzimi rilevanti per malattie, PIM1 e CDK1. Hanno addestrato il loro modello congiunto su dataset esistenti di dimensioni modeste e poi hanno ordinato i nuovi composti usando tre elementi contemporaneamente: attività prevista, incertezza del modello ed estraneità. Dopo aver acquistato e testato solo 60 molecole in saggi biochimici, hanno scoperto sette molecole con potenza a bassi micromolari, tutte strutturalmente distinte dai composti di addestramento e dai tipici inibitori delle chinasi. Le strategie che favorivano bassa estraneità — pur consentendo un certo grado di incertezza — tendevano a produrre gli hit più forti, suggerendo che prestare attenzione all’estraneità può guidare l’esplorazione verso chimica promettente ma non completamente aliena.
Cosa significa per i farmaci futuri
In termini pratici, il punteggio di estraneità dà ai modelli di machine learning per la chimica un senso incorporato di quando stanno effettuando un’eccessiva estrapolazione rispetto a ciò che conoscono. Legando questo senso alla capacità del modello di ricostruire le molecole, l’approccio riflette simultaneamente la similarità chimica e l’affidabilità della previsione. Lo studio mostra che questa metrica può rivelare shift di distribuzione che i metodi standard perdono, migliorare la prioritizzazione negli screening virtuali e aiutare a scoprire nuova materia chimica in esperimenti reali. Man mano che i cacciatori di farmaci si spingono nelle vaste e in gran parte inesplorate regioni dello spazio chimico, l’estraneità offre una bussola fondata per decidere quali predizioni audaci valga la pena credere — e testare in laboratorio.
Citazione: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
Parole chiave: apprendimento automatico molecolare, scoperta di farmaci, spazio chimico, fuori-distribuzione, screening virtuale