Clear Sky Science · de
Molekulares Deep Learning am Rande des Chemieraums
Warum klügere Wirkstoffvorhersagen wichtig sind
Die moderne Wirkstoffentdeckung stützt sich zunehmend auf künstliche Intelligenz, um riesige Chemikalienbibliotheken zu durchforsten und vielversprechende Kandidaten zu identifizieren. Doch es gibt einen Haken: Viele Machine‑Learning‑Modelle funktionieren nur gut bei Molekülen, die denen ähneln, die sie bereits gesehen haben. Werden sie gebeten, ungewöhnlichere Verbindungen zu beurteilen — genau jene, die First‑in‑class‑Medikamente werden könnten —, können die Modelle übermäßig selbstsicher und falsch liegen. Diese Studie stellt eine neue Methode vor, um zu erkennen, wann ein Modell unsicher ist, und hilft Forschenden so, sicher in bislang unerforschtes Gebiet des Chemieraums vorzustoßen.
Wenn die Karte endet
In der frühen Wirkstoffforschung suchen Wissenschaftler nach „Hits“: kleinen Molekülen, die ein biologisches Ziel wie ein krankheitsrelevantes Protein beeinflussen. Da Laborexperimente für Milliarden möglicher Moleküle unmöglich sind, werden Machine‑Learning‑Modelle mit einigen hundert bis tausend bekannten Verbindungen trainiert und dann eingesetzt, um vorherzusagen, welche neuen Kandidaten einen Test wert sind. Diese Modelle versagen jedoch häufig bei Molekülen, die sich stark von den Trainingsdaten unterscheiden — ein Problem, das als Verteilungssprung (distribution shift) bekannt ist. Bestehende Absicherungen ziehen entweder eine harte Grenze um die bekannte Region und blockieren so riskantere Moleküle, oder schätzen die Vorhersageunsicherheit auf Arten ein, die bei wirklich neuen Eingaben irreführend sein können.
Ein neues Gefühl für Unvertrautheit
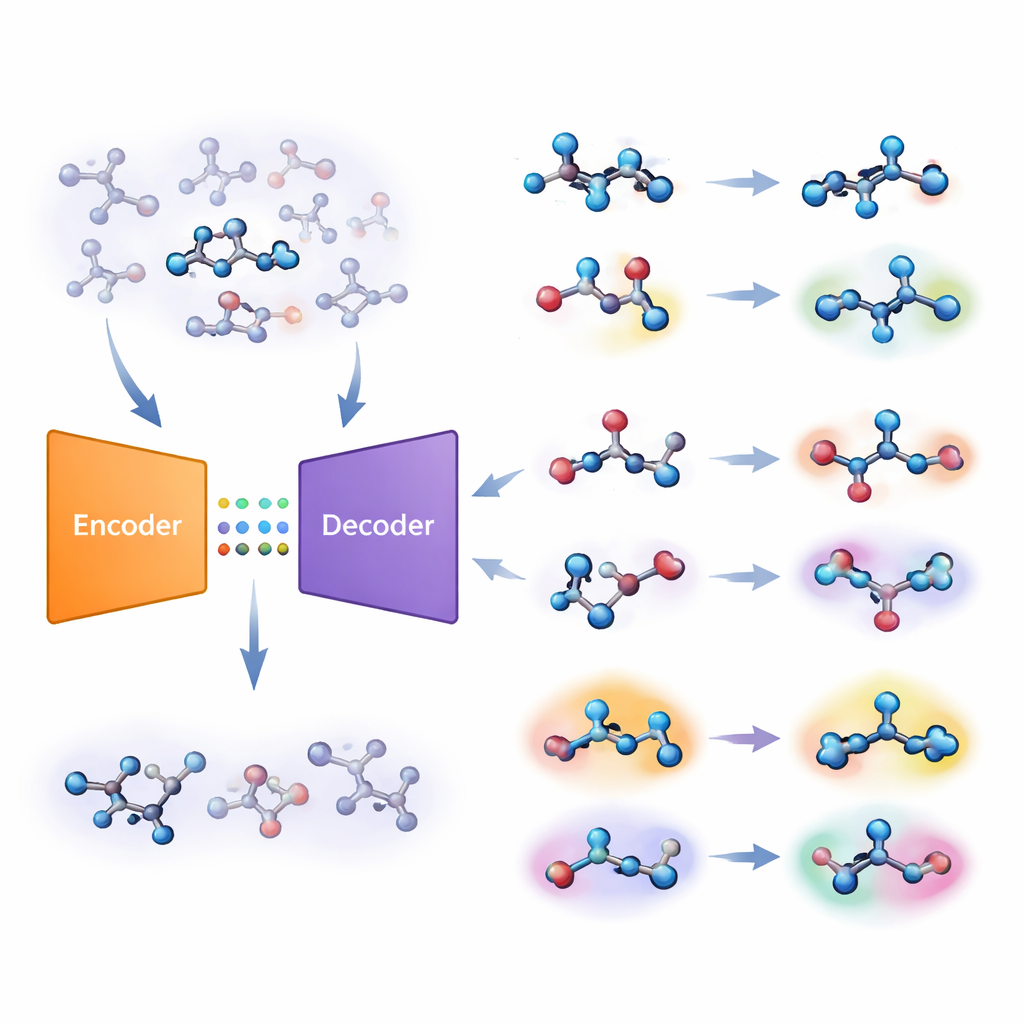
Die Autoren schlagen eine andere Strategie vor, die auf einer Art Deep‑Learning‑System namens Autoencoder beruht. Ihr „gemeinsames molekulares Modell“ lernt zwei Aufgaben gleichzeitig: vorherzusagen, ob ein Molekül an einem Ziel aktiv ist, und das Molekül aus einem komprimierten internen Code zu rekonstruieren. Gelingt dem Modell die Rekonstruktion eines bestimmten Moleküls schlecht, gilt dieses Molekül als „unvertraut“. Das Team wandelt diesen Rekonstruktionsfehler in eine numerische Kennzahl um, die sie Unvertrautheit nennen und die widerspiegelt, wie weit ein Molekül von den chemischen Mustern entfernt liegt, die das Modell tatsächlich gelernt hat. Entscheidend ist, dass dieser Wert durch das Modell‑eigene Verständnis von Chemie getrieben wird und nicht durch einfache, handgefertigte Ähnlichkeitsmaße.
Den Rand des Chemieraums testen
Um zu prüfen, wie gut Unvertrautheit erkennt, wann ein Modell an seine Grenzen stößt, stellten die Forschenden 33 Datensätze zusammen, die verschiedene biologische Ziele und Eigenschaften abdecken. Sie verwendeten Clustering‑Methoden, um jeden Datensatz in typische Beispiele und strukturell ungewöhnlichere Verbindungen zu teilen, womit sie den Unterschied zwischen gut untersuchten und neuartigen Molekülen nachbildeten. Über diese Benchmarks hinweg wiesen Moleküle, die als außerhalb der Verteilung markiert waren, durchgängig höhere Unvertrautheitswerte auf. Dieser Effekt ließ sich nicht durch triviale Merkmale wie Molekülgröße oder Komplexität erklären. Stattdessen korrelierte die Unvertrautheit eng damit, wie weit der strukturelle Kern eines Moleküls von dem der Trainingsverbindungen entfernt war, was bestätigt, dass das Modell effektiv wahrnahm, wie „ausserhalb der Karte“ ein Molekül lag.
Was alleinige Unsicherheit übersehen kann
Das Team verglich Unvertrautheit anschließend mit mehreren gängigen Methoden zur Beurteilung der Vorhersagezuverlässigkeit, darunter Modellunsicherheit und verschiedene Maßzahlen chemischer Ähnlichkeit. Sowohl Unvertrautheit als auch Unsicherheit hingen mit der Leistungsfähigkeit des Klassifikators zusammen: Wenn eine der Kennzahlen hoch war, waren Vorhersagen tendenziell weniger genau. Doch die beiden Signale waren weitgehend unabhängig. Unvertrautheit erfasste sowohl strukturelle Distanz als auch Leistung, während Unsicherheit allein Struktur nur schlecht widerspiegelte, besonders wenn Moleküle aus einer sehr unterschiedlichen Verteilung stammten. In umfangreichen virtuellen Screenings mit mehr als einer Million kommerzieller Moleküle trennte Unvertrautheit routinemäßige Verbindungen scharf von wirklich neuartigen, während Unsicherheit wenig Unterschied zwischen den beiden Gruppen andeutete.
Vom Computerbildschirm ins Nasslabor
Um die praktische Wirkung zu demonstrieren, führten die Forschenden ein prospektives Screening an etwa 180.000 käuflichen Molekülen durch und suchten Inhibitoren für zwei krankheitsrelevante Enzyme, PIM1 und CDK1. Sie trainierten ihr gemeinsames Modell mit vergleichsweise kleinen bestehenden Datensätzen und bewerteten neue Verbindungen anschließend nach drei Komponenten zugleich: vorhergesagter Aktivität, Modellunsicherheit und Unvertrautheit. Nachdem sie nur 60 Moleküle gekauft und in biochemischen Assays getestet hatten, entdeckten sie sieben Verbindungen mit niedriger mikromolarer Wirksamkeit, die alle strukturell von den Trainingsverbindungen und typischen Kinaseinhibitoren verschieden waren. Strategien, die geringe Unvertrautheit bevorzugten — bei gleichzeitiger Berücksichtigung von Unsicherheit — lieferten tendenziell die stärksten Hits, was nahelegt, dass die Beachtung von Unvertrautheit die Erkundung in Richtung vielversprechender, aber nicht völlig fremder Chemie lenken kann.
Was das für zukünftige Medikamente bedeutet
Einfach gesagt gibt die Unvertrautheitszahl Modellen für Chemie ein eingebautes Gespür dafür, wann sie zu weit vom Bekannten extrapolieren. Indem dieses Gespür an die Fähigkeit des Modells gekoppelt wird, Moleküle zu rekonstruieren, spiegelt der Ansatz zugleich chemische Ähnlichkeit und Vorhersagevertrauenswürdigkeit wider. Die Studie zeigt, dass diese Kennzahl Verteilungssprünge aufdecken kann, die Standardmethoden entgehen, die Priorisierung in virtuellen Screenings verbessert und dabei hilft, neue chemische Materie in realen Experimenten zu finden. Während Wirkstoffsuchende immer weiter in die weiten, weitgehend unerforschten Bereiche des Chemieraums vorstoßen, bietet Unvertrautheit einen prinzipiengeleiteten Kompass dafür, welche mutigen Vorhersagen glaubwürdig sind und im Labor getestet werden sollten.
Zitation: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
Schlüsselwörter: molekulares maschinelles Lernen, Wirkstoffforschung, Chemieraum, außerhalb der Verteilung, virtuelles Screening