Clear Sky Science · ru
Молекулярное глубокое обучение на грани химического пространства
Почему важны более точные предсказания лекарств
Современный поиск лекарств всё чаще опирается на искусственный интеллект, чтобы просеивать огромные библиотеки химических соединений и отмечать перспективные кандидатуры. Но есть загвоздка: многие модели машинного обучения хорошо работают лишь с молекулами, которые очень похожи на те, что они уже видели. Когда их просят оценить более необычные соединения — те самые, что могут стать препаратами первого в своём классе — модели могут стать излишне самоуверенными и ошибаться. В этом исследовании предложен новый способ определить, когда модель находится на шаткой почве, что помогает исследователям безопасно продвигаться в неизведанные области химического пространства.
Когда карта кончается
На ранних этапах поиска лекарств учёные ищут «хиты»: маленькие молекулы, воздействующие на биологическую мишень, например белок, связанный с болезнью. Так как проводить лабораторные эксперименты по миллиардам возможных молекул невозможно, модели машинного обучения обучают на нескольких сотнях или тысячах известных соединений и затем используют, чтобы предсказать, какие новые стоит тестировать. Однако эти модели склонны давать сбои на молекулах, непохожих на те, что в обучающей выборке — это проблема, известная как сдвиг распределения. Существующие предохранители либо ограничивают жёсткой границей знакомую область, блокируя более смелые молекулы, либо оценивают неопределённость предсказаний способами, которые всё ещё могут вводить в заблуждение при столкновении модели с действительно новым объектом.
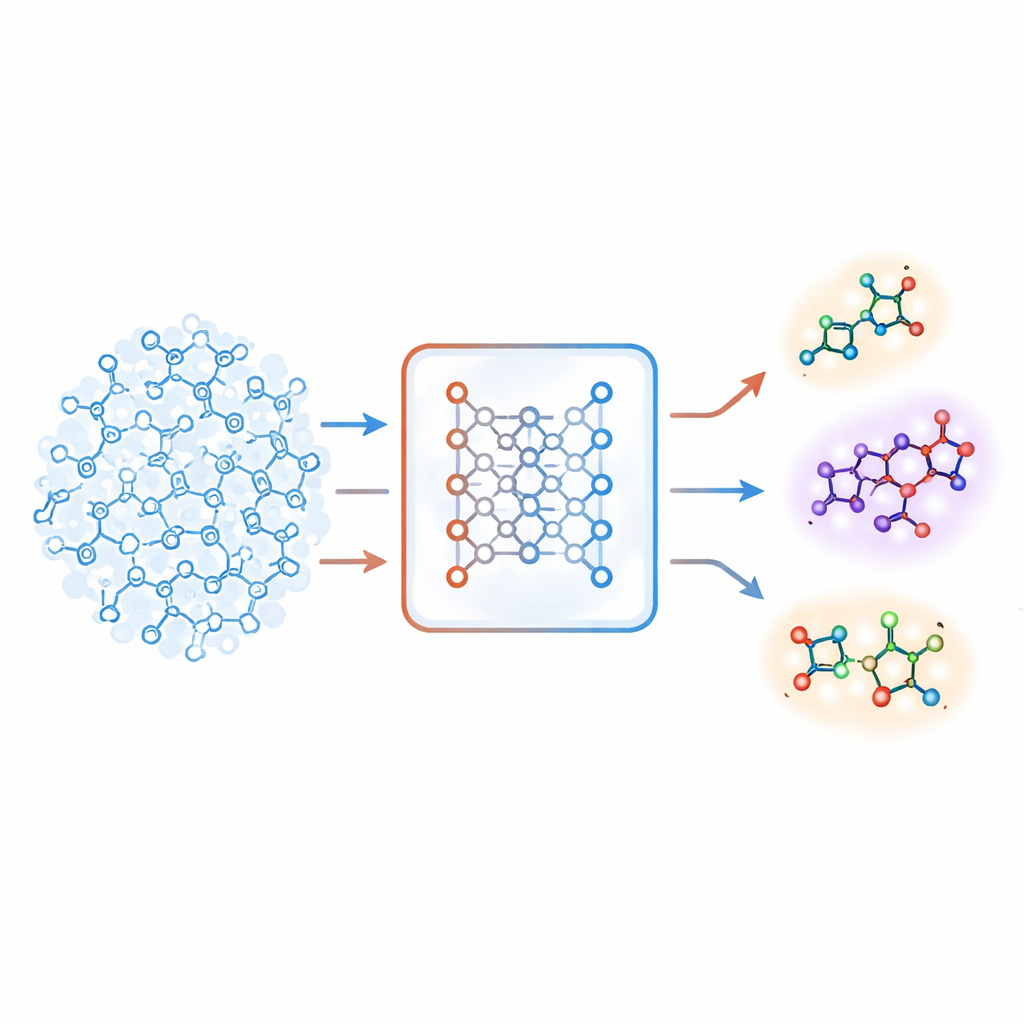
Новое чувство незнакомости
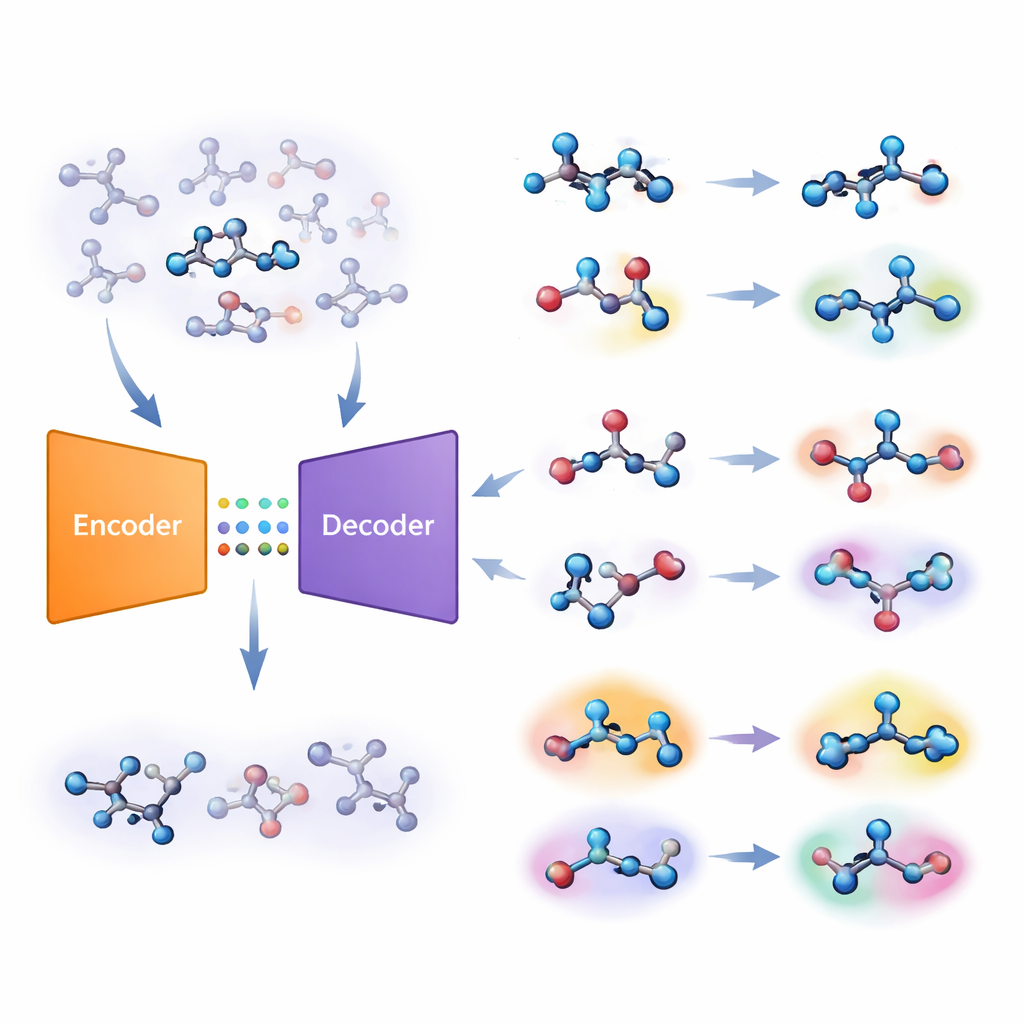
Авторы предлагают иной подход, основанный на типе систем глубокого обучения, называемом автоэнкодером. Их «совместная молекулярная модель» обучается решать две задачи одновременно: предсказывать, активна ли молекула на заданную мишень, и восстанавливать молекулу из сжатого внутреннего кода. Если модели сложно точно восстановить конкретную молекулу, эта молекула считается «незнакомой». Команда переводит ошибку реконструкции в числовой показатель — незнакомость — который отражает, насколько далеко молекула лежит от химических закономерностей, фактически выученных моделью. Ключевое: этот показатель определяется собственным пониманием модели химии, а не простыми вручную созданными мерами сходства.
Проверка границ химического пространства
Чтобы проверить, насколько хорошо незнакомость обнаруживает моменты, когда модель выходит за рамки своих возможностей, исследователи собрали 33 набора данных, охватывающие разные биологические мишени и свойства. Они использовали методы кластеризации, чтобы разделить каждый набор на типичные примеры и более структурно необычные, имитируя разницу между хорошо изученными и новыми молекулами. Во всех этих бенчмарках молекулы, помеченные как «вне распределения», последовательно имели более высокие показатели незнакомости. Этот эффект нельзя было объяснить тривиальными признаками, такими как размер или сложность молекулы. Вместо этого незнакомость тесно соответствовала тому, насколько далеко структурное ядро молекулы удалено от ядра обучающих соединений, подтверждая, что модель эффективно улавливает, насколько «вне карты» находится молекула.
То, что одна только неопределённость может пропустить
Далее команда сравнила незнакомость с несколькими распространёнными способами оценки надёжности предсказаний, включая неопределённость модели и разные меры химического сходства. И незнакомость, и неопределённость были связаны с тем, как хорошо классификатор работает: при высоких значениях любой из метрик предсказания, как правило, становились менее точными. Однако эти два сигнала были в значительной степени независимы. Незнакомость улавливала и структурное расстояние, и ухудшение качества предсказаний, в то время как одна лишь неопределённость плохо отражала структуру, особенно когда молекулы происходили из весьма отличного распределения. В масштабных виртуальных скринингах более миллиона коммерчески доступных молекул незнакомость чётко разделяла рутинные соединения и действительно новые, тогда как неопределённость почти не показывала различий между этими группами.
От экрана компьютера к влажной лаборатории
Чтобы продемонстрировать практический эффект, исследователи провели перспективный скрининг примерно 180 000 доступных к покупке молекул в поисках ингибиторов двух ферментов, имеющих отношение к болезни, PIM1 и CDK1. Они обучили свою совместную модель на умеренных существующих наборах данных и затем ранжировали новые соединения, используя три компонента одновременно: прогнозируемую активность, неопределённость модели и незнакомость. После покупки и тестирования всего 60 молекул в биохимических анализах они обнаружили семь с низкой микромолярной активностью, все структурно отличные от обучающих соединений и типичных ингибиторов киназ. Стратегии, отдававшие приоритет низкой незнакомости — при сохранении допуска неопределённости — как правило, давали самые сильные хиты, что указывает на то, что внимание к незнакомости может направлять исследование в сторону многообещающей, но не полностью чуждой химии.
Что это значит для будущих лекарств
Проще говоря, показатель незнакомости даёт моделям машинного обучения для химии встроенное чувство того, когда они экстраполируют слишком далеко от того, что знают. Связывая это чувство со способностью модели восстанавливать молекулы, подход одновременно отражает химическое сходство и надёжность предсказаний. Исследование показывает, что эта метрика может выявлять сдвиги распределения, которые стандартные методы пропускают, улучшать приоритизацию в виртуальных скринингах и помогать обнаруживать новые химические структуры в реальных экспериментах. По мере того как охотники за лекарствами всё глубже проникают в обширные, в значительной степени неисследованные просторы химического пространства, незнакомость предлагает принципиальный компас для решения, каким смелым предсказаниям стоит верить — и проверять в лаборатории.
Цитирование: van Tilborg, D., Rossen, L. & Grisoni, F. Molecular deep learning at the edge of chemical space. Nat Mach Intell 8, 575–587 (2026). https://doi.org/10.1038/s42256-026-01216-w
Ключевые слова: молекулярное машинное обучение, поиск лекарств, химическое пространство, вне распределения, виртуальный скрининг