Clear Sky Science · zh
具有多体消息传递的等变电子哈密顿量预测
为什么更快地预测电子很重要
设计新电池、计算机芯片和量子器件往往依赖于理解电子如何在材料中运动。衡量这一点的金标准是名为密度泛函理论的量子方法,它精确但在大型或复杂体系中极其缓慢。本文提出了一种新的机器学习模型 MACE‑H,它可以以显著的准确性模拟这些昂贵的电子量子计算,但成本只是其一小部分。对非专业读者而言,这项工作指向在任何人切割金属或生长晶体之前对先进材料进行更快速的数字筛选。
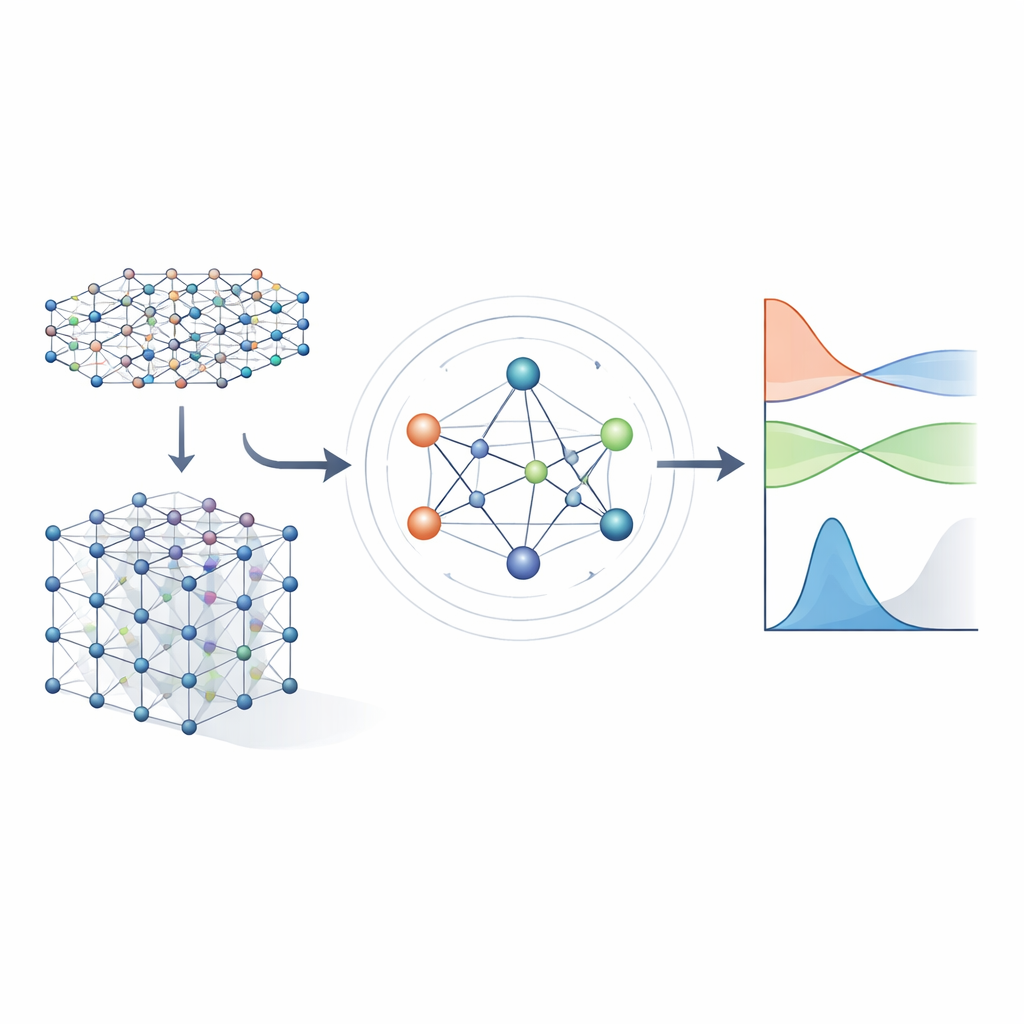
从繁重方程到学习到的捷径
传统电子结构方法通过求解称为哈密顿量的大型数学对象来工作,该对象编码了电子与原子核及相互之间的相互作用。对于现实材料,哈密顿量以一个巨大的矩阵表示,随着体系增大其尺寸呈爆炸性增长,使得直接计算代价越来越高。早期的机器学习方法试图通过使用近似的紧束缚模型或仅预测能量等标量性质来简化问题。这些方法可以很快,但通常无法保留足够的电子结构细节,因此难以在多种材料间可靠地预测能带结构、输运或光学行为。
尊重对称性的神经网络
作者基于新一代明确尊重三维空间对称性的神经网络:旋转、反射和平移。在 MACE‑H 中,材料中的原子被视为图中的节点,模型沿着它们之间的键传递“信息”。关键在于,这些消息被设计为如果你旋转或移动整个晶体,内部特征会以与基本物理相同的方式旋转和移动。这通过将信息仔细分解为在旋转下像向量和更高阶对象那样变换的分量来实现。通过这样做,模型自然处理具有不同形状的原子轨道,包括对重元素和自旋轨道效应重要的更复杂轨道。
捕捉多体化学,而不仅仅是成对作用
大多数早期的哈密顿量学习网络只允许信息沿简单的原子成对连接流动。MACE‑H 更进一步,通过引入多体消息传递:它可以以受控方式组合来自三元组及更大原子组的信息。一个特殊的节点度展开模块高效构建更高阶角特征,而不会压垮内存和计算资源。这使模型能够表示局部化学环境中的微妙模式,例如扭曲二维材料中相邻层的取向或体相金的复杂键合。同时,一个额外的边更新阶段将这些丰富的原子特征转换为每个连接一对原子轨道的哈密顿量矩阵块的预测。
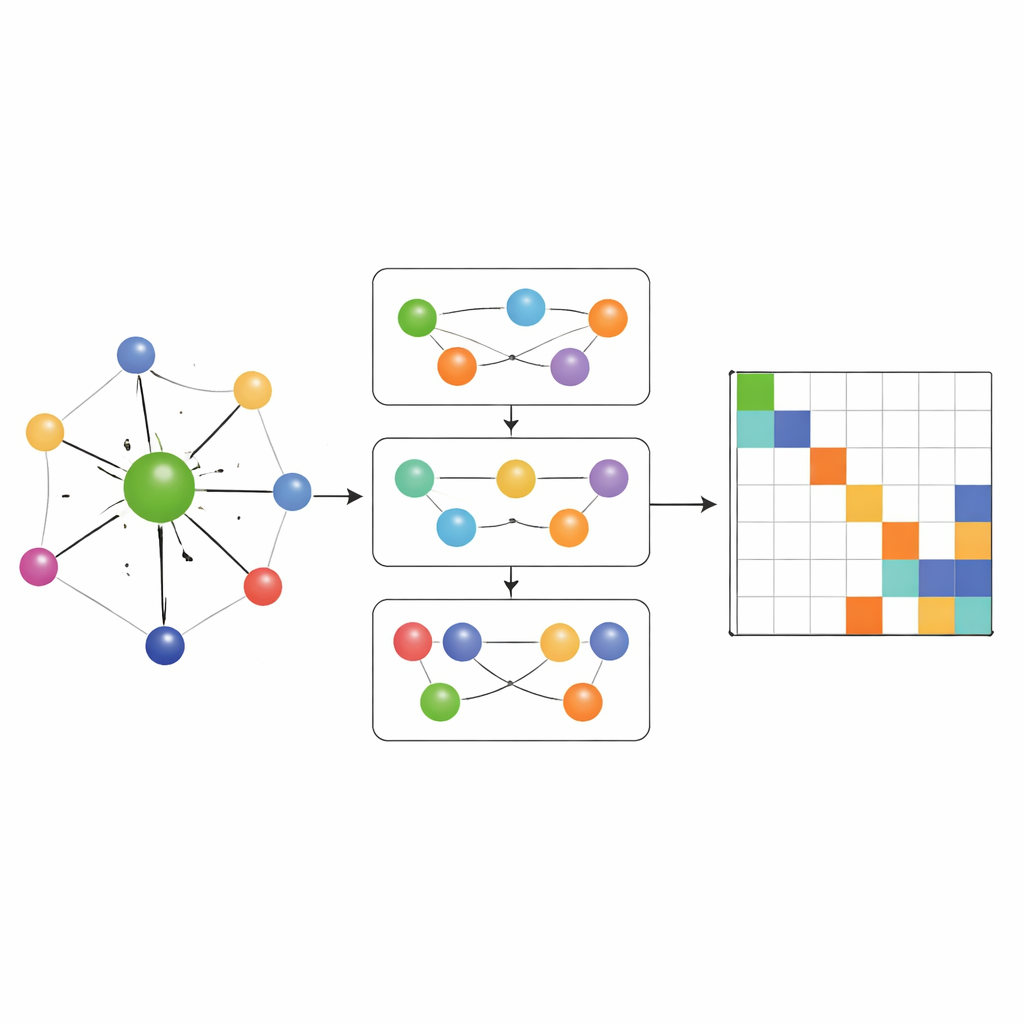
准确性、效率与智能误差检查
研究人员在若干具有挑战性的体系上测试了 MACE‑H,包括铋碲的平移与扭曲双层以及体相金的全电子计算。在这些案例中,模型对单个哈密顿量矩阵元的平均误差低于千分之一电子伏特,产生的能带结构和态密度在视觉上与完整量子力学计算无法区分。与仅使用成对消息的强基线模型相比,MACE‑H 始终更精确,并且达到给定误差水平所需的训练数据更少,同时在体系规模上保持近线性扩展。该架构倾向于强烈聚焦于附近的原子环境,这提高了数据效率,但略微降低了对极长程结构变化的敏感性;然而,即使在那些具有挑战性的扭曲结构中,费米能级附近的电子性质仍能很好地再现。作者还表明,当哈密顿量的不同部分跨越多个数量级变化时,精心设计的“移位与缩放”步骤能稳定训练,并建议使用预测矩阵是否满足基本对称性(厄米性)作为一种快速、无标签的可靠性指示器。
迈向快速的材料发现
简而言之,MACE‑H 学会模拟用于电子的复杂量子力学求解器,同时保留产生关键电子性能的完整矩阵结构。由于其准确、数据高效且可扩展,它可以插入现有电子结构代码以加速能带计算,指导候选材料的高通量筛选,或帮助耦合电子运动与原子运动的模拟。该方法具有推广到其他量子算符(例如密度矩阵)的通用性,为更快速的自洽计算开辟了一条途径。随着此类模型成熟并获得稳健的不确定性估计,它们很可能成为虚拟发现和设计新电子材料的核心工具。
引用: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
关键词: 机器学习 哈密顿量, 图神经网络, 电子结构, 材料发现, 密度泛函理论