Clear Sky Science · it
Predizione equivariant dell'Hamiltoniana elettronica con message passing many‑body
Perché è importante prevedere gli elettroni più rapidamente
Progettare nuove batterie, chip per computer e dispositivi quantistici spesso dipende dalla comprensione di come gli elettroni si muovono attraverso un materiale. Lo standard di riferimento per questo è un metodo quantistico chiamato teoria del funzionale della densità, che è accurato ma dolorosamente lento per sistemi grandi o complessi. Questo articolo introduce un nuovo modello di machine learning, MACE‑H, che è in grado di imitare questi costosi calcoli quantistici sugli elettroni con notevole accuratezza, ma a una frazione del costo. Per i non specialisti, questo lavoro indica la possibilità di screening digitale molto più rapidi di materiali avanzati prima che chiunque tagli il metallo o cresca un cristallo.
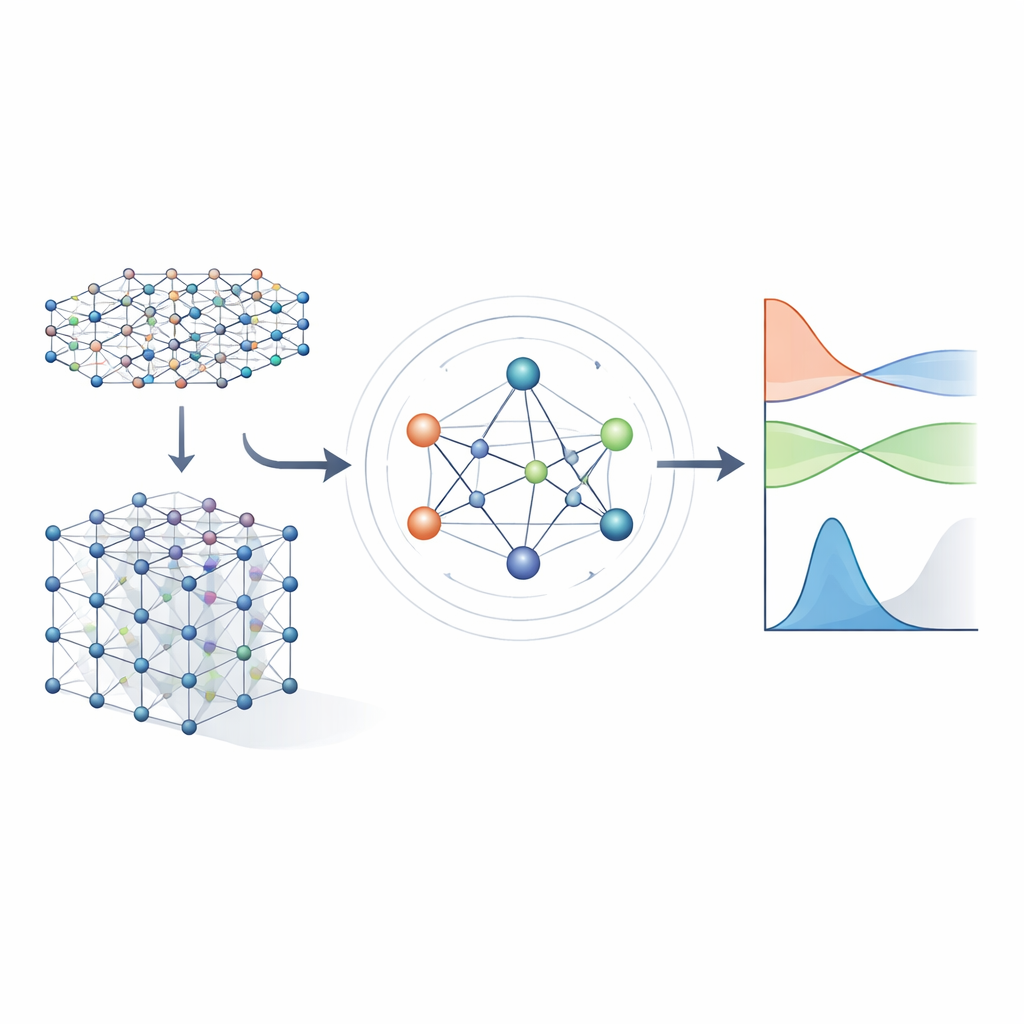
Dalle equazioni pesanti a scorciatoie apprese
I metodi tradizionali di struttura elettronica operano risolvendo un grande oggetto matematico chiamato Hamiltoniana, che codifica come gli elettroni interagiscono con i nuclei atomici e tra di loro. Per materiali realistici, l'Hamiltoniana è rappresentata come una matrice enorme la cui dimensione esplode con la crescita del sistema, rendendo il calcolo diretto sempre più costoso. Approcci di machine learning precedenti cercavano di semplificare il problema usando modelli tight‑binding approssimati o predicendo solo proprietà scalari come le energie. Questi approcci possono essere veloci, ma di solito non conservano abbastanza dettaglio sulla struttura elettronica per prevedere in modo affidabile bande, trasporto o comportamento ottico attraverso molti materiali diversi.
Una rete neurale che rispetta la simmetria
Gli autori si basano su una nuova generazione di reti neurali che rispettano esplicitamente le simmetrie dello spazio tridimensionale: rotazioni, riflessioni e traslazioni. In MACE‑H, gli atomi in un materiale sono trattati come nodi di un grafo e il modello passa “messaggi” lungo i legami tra di essi. Fondamentalmente, questi messaggi sono progettati in modo tale che se si ruota o si sposta l'intero cristallo, le caratteristiche interne ruotino e si spostino esattamente nello stesso modo previsto dalla fisica sottostante. Ciò si ottiene tramite una decomposizione accurata delle informazioni in componenti che si trasformano come vettori e oggetti di ordine superiore sotto rotazione. Così facendo, il modello gestisce in modo naturale gli orbitali atomici di diversa forma, inclusi quelli più complessi importanti per elementi pesanti e per gli effetti spin‑orbitale.
Catturare la chimica many‑body, non solo le coppie
La maggior parte delle reti che apprendono l'Hamiltoniana in precedenza consentivano il flusso di informazioni solo lungo semplici connessioni a due corpi tra atomi. MACE‑H va oltre incorporando message passing many‑body: può combinare informazioni da triplette e gruppi più grandi di atomi in modo controllato. Un modulo speciale di espansione in funzione del grado del nodo costruisce in modo efficiente caratteristiche angolari di ordine superiore senza sovraccaricare la memoria e le risorse di calcolo. Questo permette al modello di rappresentare schemi sottili nell'ambiente chimico locale, come l'orientazione degli strati vicini in materiali bidimensionali ritorti o il legame complesso nell'oro massiccio. Allo stesso tempo, una fase aggiuntiva di aggiornamento degli archi converte queste ricche caratteristiche atomiche in predizioni per ciascun blocco della matrice Hamiltoniana che collega coppie di orbitali atomici.
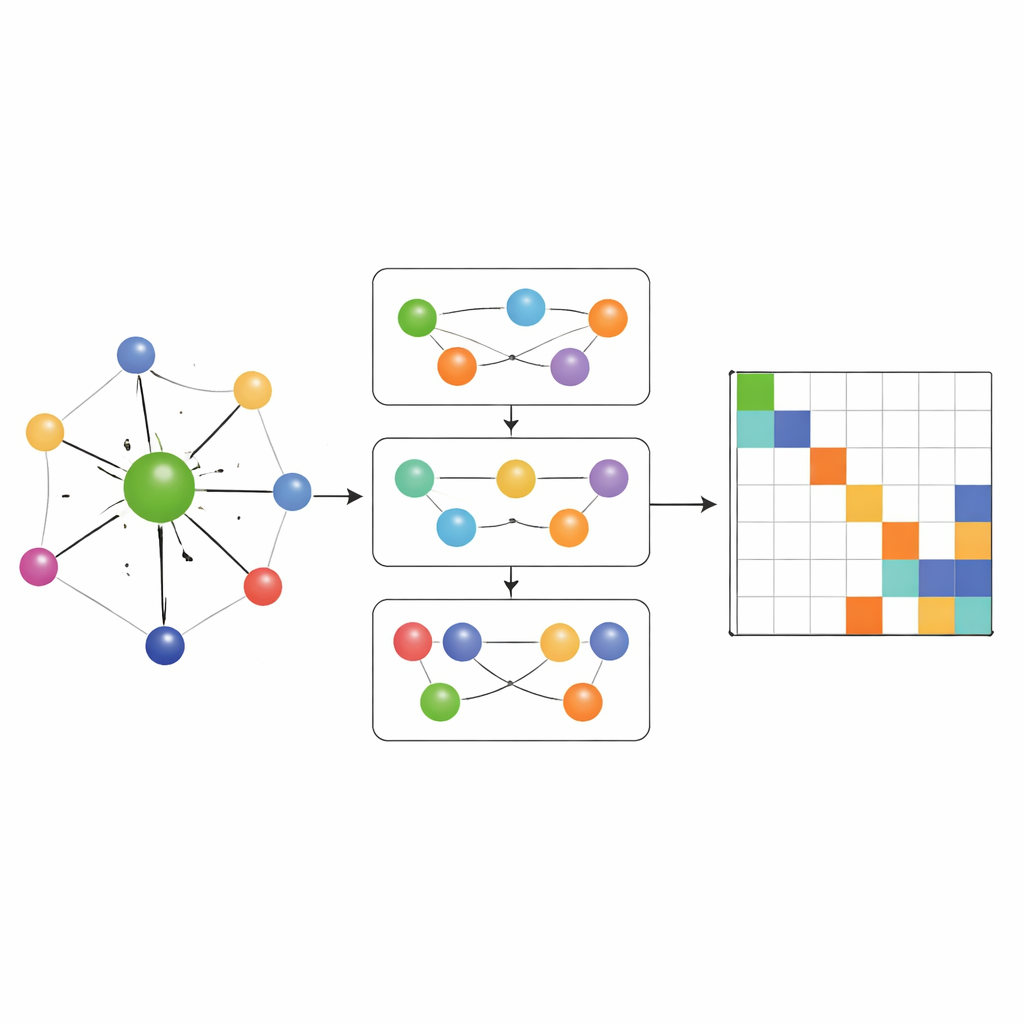
Accuratezza, efficienza e controlli intelligenti degli errori
I ricercatori testano MACE‑H su diversi sistemi impegnativi, inclusi bilayer scostati e ritorti di tellururo di bismuto e calcoli all‑electron per l'oro massiccio. In questi casi, il modello predice elementi individuali della matrice Hamiltoniana con errori medi inferiori a un millesimo di elettronvolt, e le conseguenti strutture di banda e densità di stati sono visivamente indistinguibili dai calcoli quantomeccanici completi. Rispetto a un forte modello precedente che usa solo messaggi a coppie, MACE‑H è costantemente più accurato e richiede meno dati di addestramento per raggiungere un dato livello di errore, mantenendo una scalabilità quasi lineare con la dimensione del sistema. L'architettura tende a concentrarsi fortemente sull'ambiente atomico vicino, il che aumenta l'efficienza dei dati ma riduce leggermente la sensibilità a cambiamenti strutturali molto a lungo raggio; tuttavia, anche in quelle strutture ritorte sfidanti, le proprietà elettroniche vicino al livello di Fermi restano ben riprodotte. Gli autori mostrano inoltre che una fase di “shift‑and‑scale” accuratamente progettata stabilizza l'addestramento quando diverse parti dell'Hamiltoniana variano su molteplici ordini di grandezza, e propongono di usare quanto bene la matrice predetta rispetta una simmetria di base (l'ermiticità) come indicatore rapido e senza etichette di affidabilità.
Verso una rapida scoperta di materiali
In termini semplici, MACE‑H impara a emulare un costoso risolutore quantomeccanico per gli elettroni pur mantenendo la struttura matriciale completa che sta alla base delle proprietà elettroniche chiave. Poiché è accurato, efficiente in termini di dati e scalabile, può essere integrato nei codici di struttura elettronica esistenti per accelerare i calcoli delle bande, guidare lo screening ad alto rendimento di materiali candidati o aiutare simulazioni che accoppiano il moto elettronico al moto atomico. L'approccio è sufficientemente generale da poter essere esteso ad altri operatori quantistici, come le matrici di densità, aprendo una via a calcoli auto‑consistenti più rapidi. Man mano che questi modelli maturano e acquisiscono stime robuste di incertezza, è probabile che diventino strumenti centrali nella scoperta e progettazione virtuale di nuovi materiali elettronici.
Citazione: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Parole chiave: Hamiltoniana tramite machine learning, reti neurali a grafo, struttura elettronica, scoperta di materiali, teoria del funzionale della densità