Clear Sky Science · de
Äquivarianten Vorhersage elektronischer Hamiltonoperatoren mit Many‑Body‑Message‑Passing
Warum schnellere Elektronenvorhersagen wichtig sind
Die Entwicklung neuer Batterien, Computerchips und Quantenbauteile hängt oft davon ab, wie Elektronen sich in einem Material bewegen. Der Goldstandard dafür ist eine Quantenmethode namens Dichtfunktionaltheorie, die zwar genau ist, aber bei großen oder komplexen Systemen quälend langsam arbeitet. Dieses Paper stellt ein neues maschinelles Lernmodell vor, MACE‑H, das diese aufwändigen Quantenberechnungen für Elektronen mit bemerkenswerter Genauigkeit nachahmen kann — jedoch zu einem Bruchteil der Kosten. Für Nicht‑Spezialisten weist diese Arbeit den Weg zu schnelleren digitalen Durchsuchungen fortschrittlicher Materialien, bevor überhaupt Metall geschnitten oder ein Kristall gezogen wird.
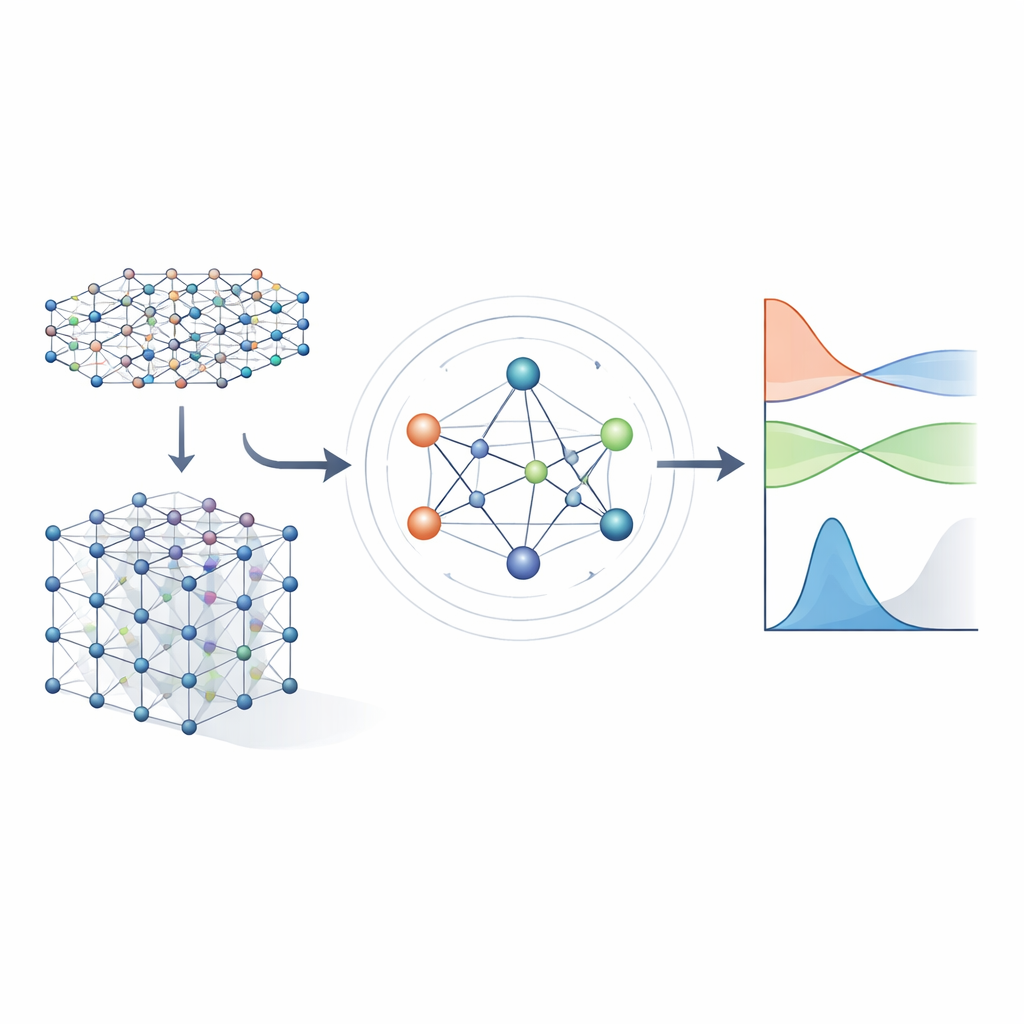
Von schweren Gleichungen zu gelernten Abkürzungen
Traditionelle Methoden der Elektronenstruktur arbeiten, indem sie ein großes mathematisches Objekt lösen, den Hamiltonoperator, der kodiert, wie Elektronen mit Atomkernen und miteinander wechselwirken. Für realistische Materialien wird der Hamiltonoperator als riesige Matrix dargestellt, deren Größe mit dem Systemumfang explodiert, wodurch direkte Berechnungen zunehmend teuer werden. Frühere Ansätze mit maschinellem Lernen versuchten, das Problem zu vereinfachen, indem sie entweder approximative Tight‑Binding‑Modelle verwendeten oder nur skalare Eigenschaften wie Energien vorhersagten. Diese Methoden können schnell sein, behalten aber meist nicht genug Details der Elektronenstruktur bei, um zuverlässig Bandstrukturen, Transport‑ oder optische Eigenschaften über viele verschiedene Materialien hinweg vorherzusagen.
Ein neuronales Netzwerk, das Symmetrien respektiert
Die Autoren bauen auf einer neuen Generation von neuronalen Netzen auf, die explizit die Symmetrien des dreidimensionalen Raums respektieren: Rotation, Spiegelung und Translation. In MACE‑H werden Atome in einem Material als Knoten in einem Graphen behandelt, und das Modell leitet „Nachrichten“ entlang der Verbindungen zwischen ihnen. Entscheidend ist, dass diese Nachrichten so konstruiert sind, dass sich die internen Merkmale bei Rotation oder Verschiebung des gesamten Kristalls genau so transformieren, wie es die zugrunde liegende Physik vorschreibt. Dies wird durch eine sorgfältige Zerlegung der Informationen in Komponenten erreicht, die sich unter Rotation wie Vektoren und höherwertige Objekte verhalten. Dadurch kann das Modell atomare Orbitale mit unterschiedlichen Formen natürlich verarbeiten, einschließlich der komplexeren, die für schwere Elemente und Spin‑Bahneffekte wichtig sind.
Viele‑Körper‑Chemie erfassen, nicht nur Paare
Die meisten früheren Hamilton‑Lernnetzwerke erlaubten nur Informationsfluss entlang einfacher paarweiser Verbindungen zwischen Atomen. MACE‑H geht darüber hinaus, indem es Many‑Body‑Message‑Passing einbezieht: Es kann Informationen aus Tripeln und größeren Atomgruppen auf kontrollierte Weise kombinieren. Ein spezielles Modul zur Erweiterung nach Knotengrad baut effizient höherwertige Winkelmerkmale auf, ohne Speicher‑ und Rechenressourcen zu überfordern. Dadurch kann das Modell subtile Muster in der lokalen chemischen Umgebung darstellen, wie die Orientierung benachbarter Schichten in verdrehten zweidimensionalen Materialien oder die komplexe Bindung in massives Gold. Gleichzeitig wandelt eine zusätzliche Kanten‑Aktualisierungsstufe diese reichen atomaren Merkmale in Vorhersagen für jeden Block der Hamiltonmatrix um, der Paare von Atomorbitalen verbindet.
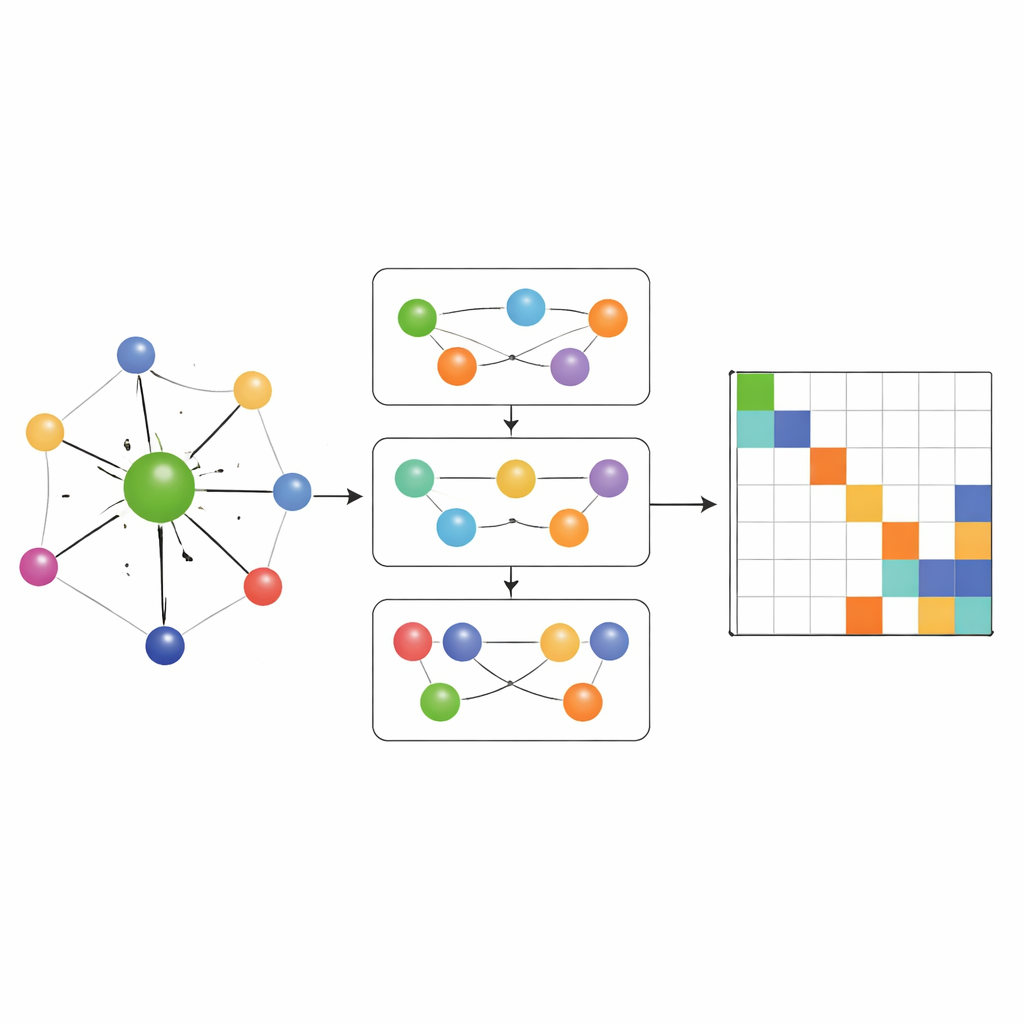
Genauigkeit, Effizienz und intelligente Fehlerkontrollen
Die Forschenden testen MACE‑H an mehreren anspruchsvollen Systemen, darunter verschobene und verdrehte Bilayer aus Wismut‑tellurid und All‑Electron‑Berechnungen für massives Gold. In diesen Fällen sagt das Modell einzelne Matrixelemente des Hamiltonoperators mit durchschnittlichen Fehlern unter einem Tausendstel Elektronenvolt voraus, und die resultierenden Bandstrukturen sowie Zustandsdichten sind visuell nicht von vollständigen quantenmechanischen Berechnungen zu unterscheiden. Im Vergleich zu einem leistungsfähigen früheren Modell, das nur paarweise Nachrichten nutzt, ist MACE‑H durchgängig genauer und benötigt weniger Trainingsdaten, um ein gegebenes Fehlerniveau zu erreichen, während es annähernd linear mit der Systemgröße skaliert. Die Architektur fokussiert stark auf die nahe Umgebung der Atome, was die Dateneffizienz steigert, aber die Sensitivität für sehr langreichweitige strukturelle Änderungen leicht reduziert; selbst in den herausfordernden verdrehten Strukturen bleiben die elektronischen Eigenschaften nahe dem Fermi‑Niveau jedoch gut reproduziert. Die Autor:innen zeigen außerdem, dass ein sorgfältig gestalteter „Shift‑and‑Scale“‑Schritt das Training stabilisiert, wenn verschiedene Teile des Hamiltonoperators über viele Größenordnungen variieren, und schlagen vor, die Einhaltung einer grundlegenden Symmetrie (Hermitizität) der vorhergesagten Matrix als schnellen, kennzeichenfreien Indikator für die Zuverlässigkeit zu verwenden.
Auf dem Weg zu schneller Materialentdeckung
Kurz gesagt, MACE‑H lernt, einen aufwändigen quantenmechanischen Löser für Elektronen zu emulieren, während es die vollständige Matrixstruktur bewahrt, die den Schlüssel zu elektronischen Eigenschaften bildet. Da es genau, daten‑effizient und skalierbar ist, lässt es sich in bestehende Elektronenstruktur‑Codes einbetten, um Bandstruktur‑Berechnungen zu beschleunigen, High‑Throughput‑Screenings von Kandidatenmaterialien zu leiten oder Simulationen zu unterstützen, die Elektronenbewegung mit Atombewegung koppeln. Der Ansatz ist allgemein genug, um auf andere Quantenoperatoren wie Dichtematrizen erweitert zu werden und eröffnet damit einen Weg zu schnelleren selbstkonsistenten Rechnungen. Wenn solche Modelle reifen und robuste Unsicherheitsabschätzungen liefern, werden sie vermutlich zu zentralen Werkzeugen in der virtuellen Entdeckung und dem Design neuer elektronischer Materialien.
Zitation: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Schlüsselwörter: maschinelles Lernen Hamiltonoperator, Graph‑Neuronale Netzwerke, Elektronenstruktur, Materialentdeckung, Dichtfunktionaltheorie