Clear Sky Science · fr
Prédiction d'Hamiltonien électronique équivariant par passage de messages many‑body
Pourquoi prédire les électrons plus rapidement importe
Concevoir de nouvelles batteries, puces informatiques et dispositifs quantiques dépend souvent de la compréhension du déplacement des électrons dans un matériau. La référence est une méthode quantique appelée théorie de la fonctionnelle de la densité, précise mais terriblement lente pour les systèmes grands ou complexes. Cet article présente un nouveau modèle d'apprentissage automatique, MACE‑H, capable d'imiter ces calculs quantiques coûteux pour les électrons avec une précision remarquable, mais à une fraction du coût. Pour les non‑spécialistes, ce travail ouvre la voie à un criblage numérique plus rapide des matériaux avancés avant toute découpe de métal ou croissance de cristal.
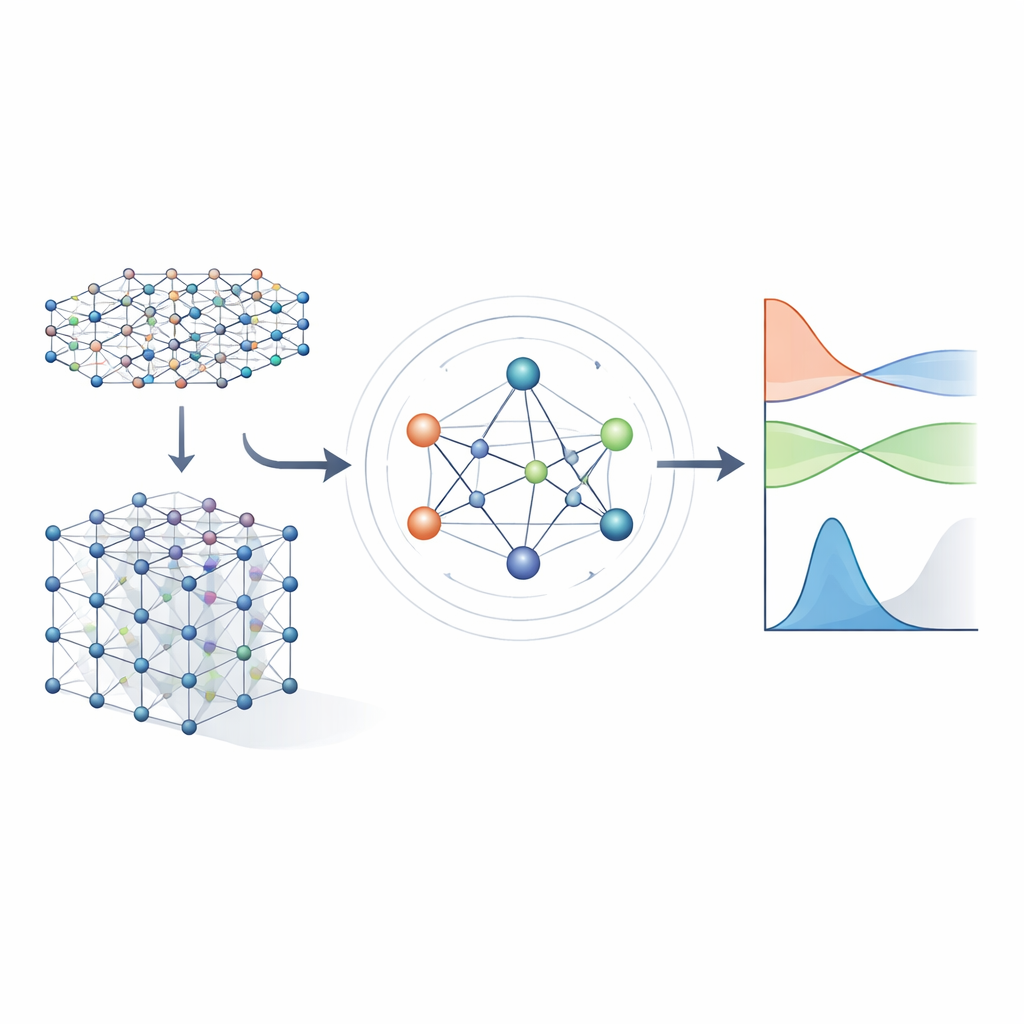
Des équations lourdes aux raccourcis appris
Les méthodes classiques de structure électronique fonctionnent en résolvant un objet mathématique volumineux appelé Hamiltonien, qui encode comment les électrons interagissent avec les noyaux atomiques et entre eux. Pour des matériaux réalistes, le Hamiltonien se représente par une énorme matrice dont la taille explose avec la croissance du système, rendant le calcul direct de plus en plus coûteux. Les approches antérieures d'apprentissage automatique tentaient de simplifier le problème soit en utilisant des modèles de liaisons serrées approximatifs, soit en prédisant seulement des grandeurs scalaires comme les énergies. Ces méthodes peuvent être rapides, mais elles conservent généralement trop peu de détails sur la structure électronique pour prédire de manière fiable les structures de bandes, le transport ou le comportement optique à travers une grande variété de matériaux.
Un réseau neuronal qui respecte la symétrie
Les auteurs s'appuient sur une nouvelle génération de réseaux neuronaux qui respectent explicitement les symétries de l'espace tridimensionnel : rotations, réflexions et translations. Dans MACE‑H, les atomes d'un matériau sont traités comme des nœuds d'un graphe, et le modèle transmet des « messages » le long des liaisons qui les relient. De manière cruciale, ces messages sont conçus de sorte que si l'on fait tourner ou déplacer l'ensemble du cristal, les caractéristiques internes tournent et se déplacent exactement comme l'impose la physique sous‑jacente. Cela s'obtient par une décomposition soignée de l'information en composantes qui se transforment comme des vecteurs et des objets d'ordre supérieur sous rotation. Ce faisant, le modèle gère naturellement des orbitales atomiques de formes différentes, y compris les plus complexes importantes pour les éléments lourds et pour les effets spin‑orbite.
Capturer la chimie many‑body, pas seulement les paires
La plupart des réseaux d'apprentissage d'Hamiltonien antérieurs autorisaient l'écoulement d'information uniquement le long de connexions pair à pair simples entre atomes. MACE‑H va plus loin en incorporant un passage de messages many‑body : il peut combiner des informations provenant de triplets et de groupes d'atomes plus larges de manière contrôlée. Un module spécial d'expansion selon le degré des nœuds construit efficacement des caractéristiques angulaires d'ordre supérieur sans submerger la mémoire et les ressources de calcul. Cela permet au modèle de représenter des motifs subtils dans l'environnement chimique local, comme l'orientation des couches voisines dans des matériaux bidimensionnels tordus ou la liaison complexe dans l'or massif. Parallèlement, une étape supplémentaire de mise à jour des arêtes convertit ces riches caractéristiques atomiques en prédictions pour chaque bloc de la matrice Hamiltonienne qui relie des paires d'orbitales atomiques.
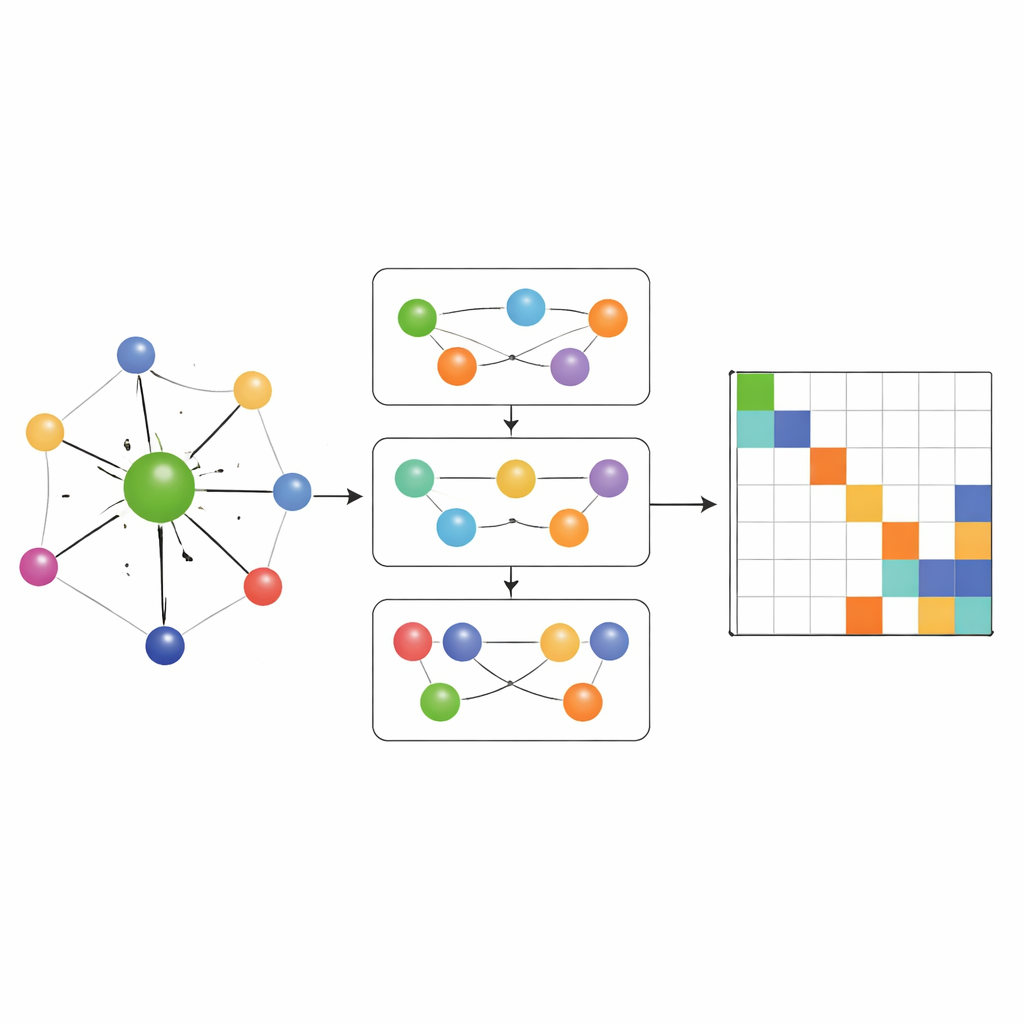
Précision, efficacité et contrôles d'erreur intelligents
Les chercheurs testent MACE‑H sur plusieurs systèmes exigeants, y compris des bilayers décalés et tordus de tellurure de bismuth et des calculs tout‑électrons pour l'or massif. Dans ces cas, le modèle prédit des éléments individuels de la matrice Hamiltonienne avec des erreurs moyennes inférieures au millième d'électron‑volt, et les structures de bandes et densités d'états résultantes sont visuellement indiscernables des calculs quantum‑mécaniques complets. Comparé à un modèle précédent performant utilisant seulement des messages pair à pair, MACE‑H est systématiquement plus précis et nécessite moins de données d'entraînement pour atteindre un niveau d'erreur donné, tout en conservant un quasi‑scaling linéaire avec la taille du système. L'architecture a tendance à se concentrer fortement sur l'environnement atomique proche, ce qui augmente l'efficacité des données mais réduit légèrement la sensibilité aux changements structuraux très longue portée ; toutefois, même dans ces structures tordues difficiles, les propriétés électroniques près du niveau de Fermi restent bien reproduites. Les auteurs montrent aussi qu'une étape soigneusement conçue de « décalage et mise à l'échelle » stabilise l'entraînement lorsque différentes parties de l'Hamiltonien varient sur de nombreux ordres de grandeur, et ils proposent d'utiliser le degré d'obéissance de la matrice prédite à une symétrie de base (l'hermiticité) comme indicateur rapide de fiabilité sans étiquettes.
Vers une découverte rapide de matériaux
En termes simples, MACE‑H apprend à émuler un solveur quantique lourd pour les électrons tout en conservant la structure matricielle complète qui sous‑tend les propriétés électroniques clés. Parce qu'il est précis, économe en données et évolutif, il peut être intégré aux codes de structure électronique existants pour accélérer les calculs de structures de bandes, guider le criblage à haut débit de matériaux candidats ou aider les simulations couplant le mouvement des électrons à celui des atomes. L'approche est suffisamment générale pour être étendue à d'autres opérateurs quantiques, comme les matrices de densité, ouvrant une voie vers des calculs auto‑consistants plus rapides. À mesure que ces modèles mûrissent et obtiennent des estimations d'incertitude robustes, ils sont susceptibles de devenir des outils centraux dans la découverte virtuelle et la conception de nouveaux matériaux électroniques.
Citation: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Mots-clés: apprentissage automatique Hamiltonien, réseaux de neurones graphes, structure électronique, découverte de matériaux, théorie de la fonctionnelle de la densité