Clear Sky Science · pt
Predição equivariante do Hamiltoniano eletrônico com passagem de mensagens many‑body
Por que prever elétrons mais rápido importa
Projetar novas baterias, chips de computador e dispositivos quânticos frequentemente depende de entender como os elétrons se movem através de um material. O padrão‑ouro para isso é um método quântico chamado teoria do funcional da densidade, que é preciso, mas dolorosamente lento para sistemas grandes ou complexos. Este artigo apresenta um novo modelo de aprendizado de máquina, MACE‑H, que pode imitar esses custosos cálculos quânticos para elétrons com precisão notável, porém a uma fração do custo. Para não especialistas, este trabalho aponta para uma triagem digital mais rápida de materiais avançados antes que alguém corte metal ou cresça um cristal.
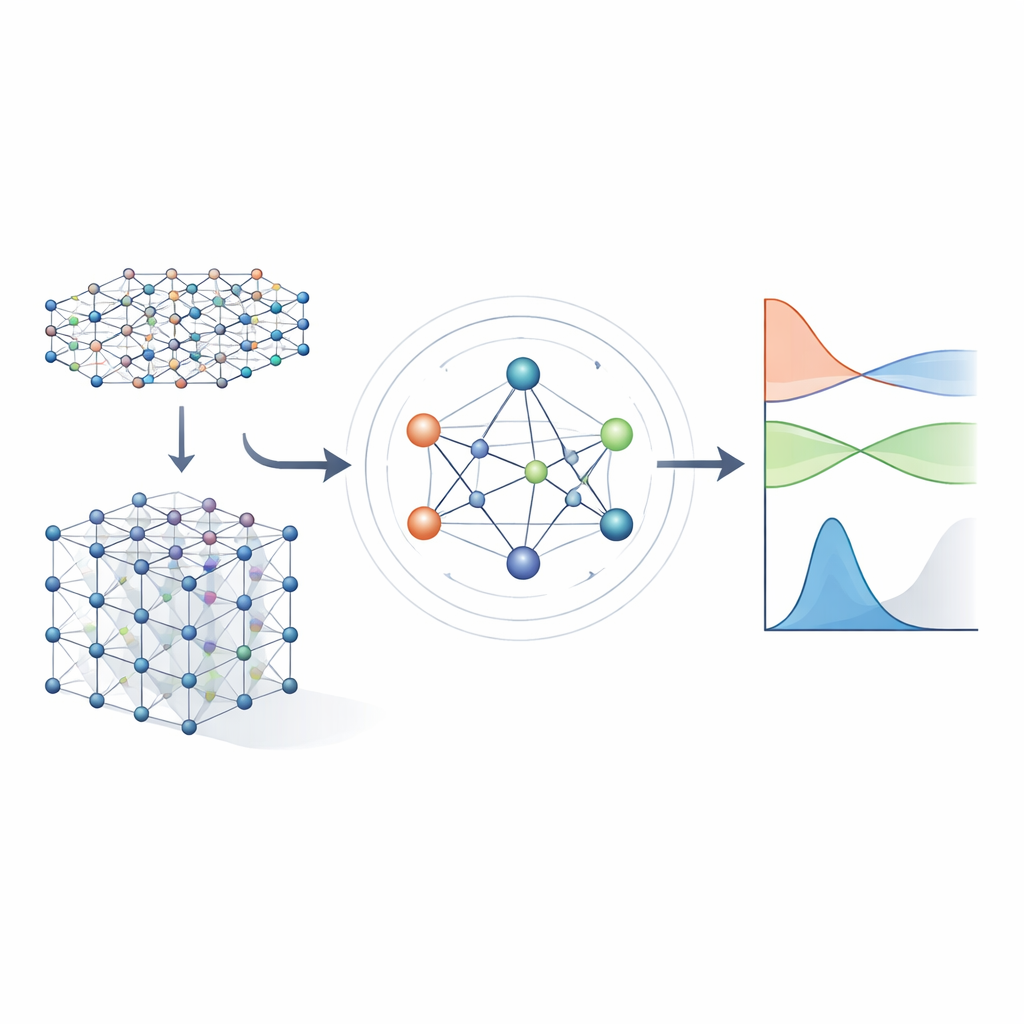
De equações pesadas a atalhos aprendidos
Métodos tradicionais de estrutura eletrônica funcionam resolvendo um grande objeto matemático chamado Hamiltoniano, que codifica como os elétrons interagem com os núcleos atômicos e entre si. Para materiais realistas, o Hamiltoniano é representado como uma enorme matriz cujo tamanho explode conforme o sistema cresce, tornando o cálculo direto cada vez mais caro. Abordagens anteriores de aprendizado de máquina tentaram simplificar o problema ou usando modelos aproximados de tight‑binding, ou prevendo apenas propriedades escalares como energias. Essas abordagens podem ser rápidas, mas geralmente não mantêm detalhes suficientes sobre a estrutura eletrônica para prever de forma confiável bandas eletrônicas, transporte ou comportamento óptico através de muitos materiais diferentes.
Uma rede neural que respeita a simetria
Os autores se baseiam em uma nova geração de redes neurais que respeitam explicitamente as simetrias do espaço tridimensional: rotações, reflexões e translações. No MACE‑H, os átomos em um material são tratados como nós em um grafo, e o modelo passa “mensagens” ao longo das ligações entre eles. Crucialmente, essas mensagens são projetadas de modo que, se você rotacionar ou deslocar todo o cristal, as características internas rotacionam e se movem exatamente da mesma forma que a física subjacente dita. Isso é alcançado por meio de uma decomposição cuidadosa da informação em componentes que se transformam como vetores e objetos de ordem superior sob rotação. Ao fazer isso, o modelo lida naturalmente com orbitais atômicos de formas diferentes, incluindo os mais complexos, importantes para elementos pesados e para efeitos de spin‑órbita.
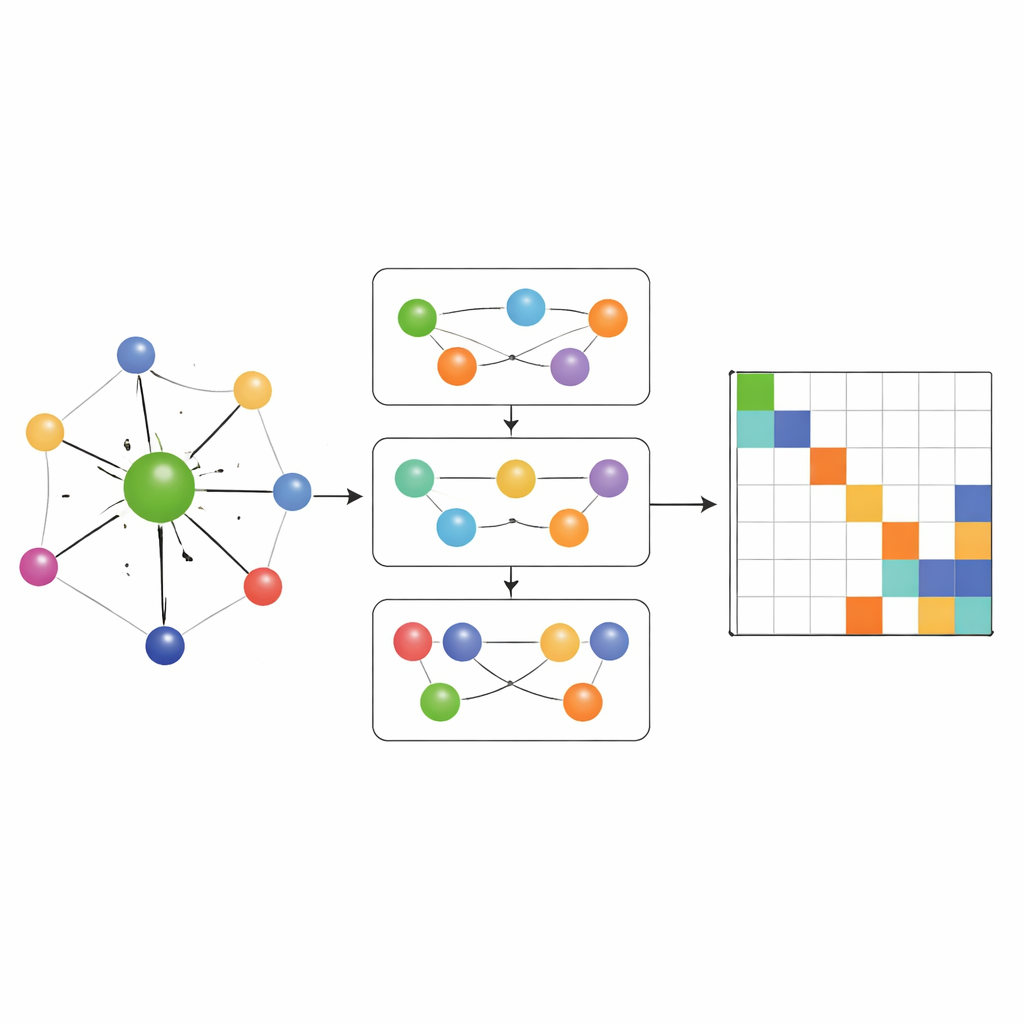
Capturando química many‑body, não apenas pares
A maioria das redes anteriores que aprendem Hamiltonianos permitia apenas que a informação fluísse por conexões pareadas simples entre átomos. O MACE‑H vai além, incorporando passagem de mensagens many‑body: ele pode combinar informação de trios e grupos maiores de átomos de forma controlada. Um módulo especial de expansão segundo o grau do nó constrói eficientemente características angulares de ordem superior sem sobrecarregar memória e recursos de computação. Isso permite que o modelo represente padrões sutis no ambiente químico local, como a orientação de camadas vizinhas em materiais bidimensionais torcidos ou as ligações complexas no ouro maciço. Ao mesmo tempo, uma etapa adicional de atualização de arestas converte essas ricas características atômicas em previsões para cada bloco da matriz do Hamiltoniano que liga pares de orbitais atômicos.
Precisão, eficiência e verificações inteligentes de erro
Os pesquisadores testam o MACE‑H em vários sistemas exigentes, incluindo biestratos deslocados e torcidos de telureto de bismuto e cálculos com todos os elétrons para ouro em bloco. Nesses casos, o modelo prevê elementos individuais da matriz do Hamiltoniano com erros médios abaixo de um milésimo de elétron‑volt, e as estruturas de bandas e densidades de estados resultantes são visualmente indistinguíveis de cálculos quântico‑mecânicos completos. Em comparação com um modelo anterior robusto que usa apenas mensagens pareadas, o MACE‑H é consistentemente mais preciso e necessita de menos dados de treinamento para atingir um dado nível de erro, mantendo escalamento quase linear com o tamanho do sistema. A arquitetura tende a focar fortemente no ambiente atômico próximo, o que aumenta a eficiência de dados, mas reduz ligeiramente a sensibilidade a mudanças estruturais de muito longo alcance; no entanto, mesmo nessas estruturas torcidas desafiadoras, as propriedades eletrônicas próximas ao nível de Fermi permanecem bem reproduzidas. Os autores também mostram que uma etapa cuidadosamente projetada de “deslocar e escalar” estabiliza o treinamento quando diferentes partes do Hamiltoniano variam por muitas ordens de grandeza, e propõem usar quão bem a matriz prevista obedece a uma simetria básica (hermiticidade) como um indicador rápido e sem rótulo de confiabilidade.
Rumo à descoberta rápida de materiais
Em termos simples, o MACE‑H aprende a emular um solucionador quântico‑mecânico pesado para elétrons enquanto acompanha a estrutura matricial completa que fundamenta propriedades eletrônicas chave. Por ser preciso, eficiente em dados e escalável, pode ser integrado a códigos de estrutura eletrônica existentes para acelerar cálculos de bandas, orientar triagens em alta vazão de materiais candidatos ou auxiliar simulações que acoplam o movimento eletrônico ao movimento atômico. A abordagem é suficientemente geral para ser estendida a outros operadores quânticos, como matrizes densidade, abrindo uma rota para cálculos autorconsistentes mais rápidos. À medida que tais modelos amadurecem e ganham estimativas robustas de incerteza, é provável que se tornem ferramentas centrais na descoberta virtual e no projeto de novos materiais eletrônicos.
Citação: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Palavras-chave: Hamiltoniano por aprendizado de máquina, redes neurais em grafos, estrutura eletrônica, descoberta de materiais, teoria do funcional da densidade