Clear Sky Science · en
Equivariant electronic Hamiltonian prediction with many-body message passing
Why predicting electrons faster matters
Designing new batteries, computer chips, and quantum devices often hinges on understanding how electrons move through a material. The gold standard for this is a quantum method called density functional theory, which is accurate but painfully slow for large or complex systems. This paper introduces a new machine‑learning model, MACE‑H, that can mimic these expensive quantum calculations for electrons with remarkable accuracy, but at a fraction of the cost. For non‑specialists, this work points toward faster digital screening of advanced materials before anyone cuts metal or grows a crystal.
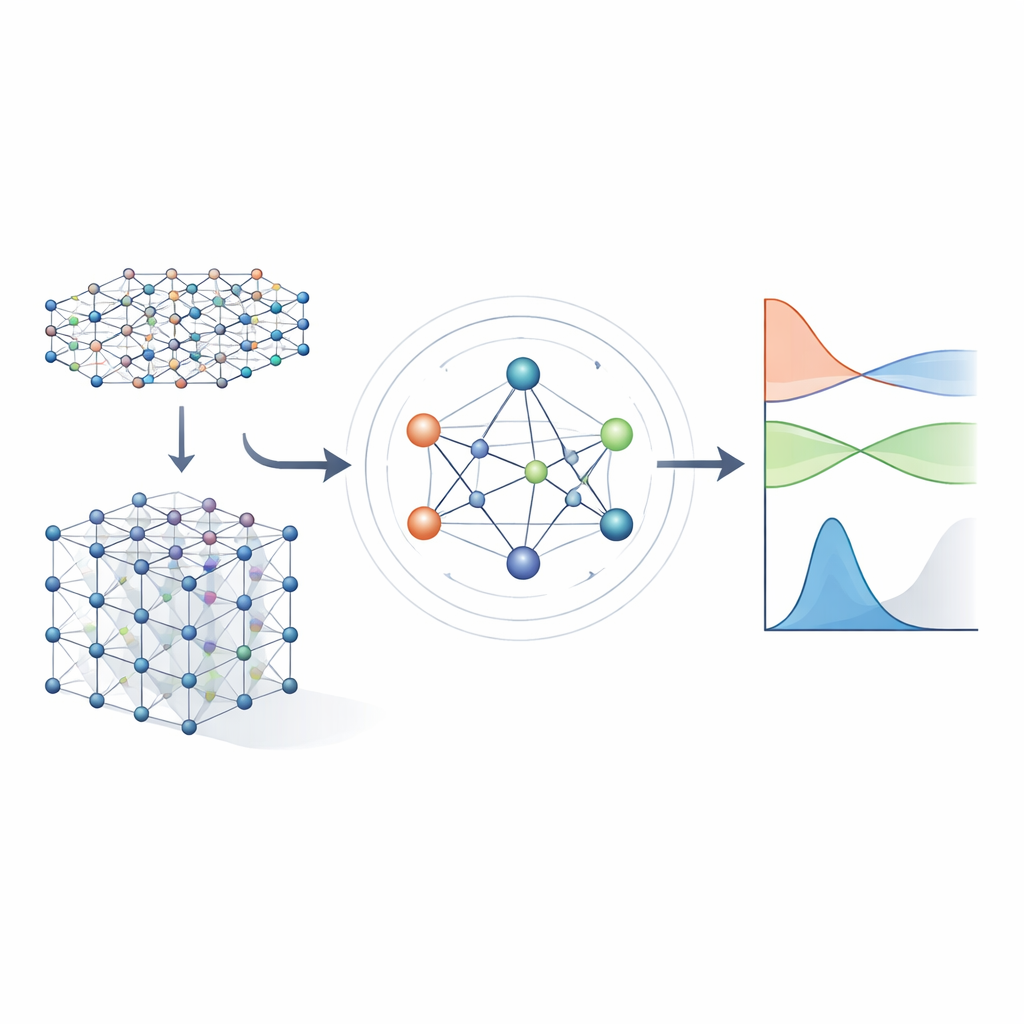
From heavy equations to learned shortcuts
Traditional electronic‑structure methods work by solving a large mathematical object called the Hamiltonian, which encodes how electrons interact with atomic nuclei and with each other. For realistic materials, the Hamiltonian is represented as a huge matrix whose size explodes as the system grows, making direct calculation increasingly expensive. Earlier machine‑learning approaches tried to simplify the problem either by using approximate tight‑binding models or by predicting only scalar properties such as energies. These approaches can be fast, but they usually do not retain enough detail about the electronic structure to reliably predict band structures, transport, or optical behavior across many different materials.
A neural network that respects symmetry
The authors build on a new generation of neural networks that explicitly respect the symmetries of three‑dimensional space: rotations, reflections, and translations. In MACE‑H, atoms in a material are treated as nodes in a graph, and the model passes "messages" along the bonds between them. Crucially, these messages are designed so that if you rotate or move the entire crystal, the internal features rotate and move in exactly the same way as the underlying physics dictates. This is achieved through a careful decomposition of the information into components that transform like vectors and higher‑order objects under rotation. By doing so, the model naturally handles atomic orbitals with different shapes, including the more complex ones that are important for heavy elements and for spin–orbit effects.
Capturing many‑body chemistry, not just pairs
Most earlier Hamiltonian‑learning networks only allowed information to flow along simple pairwise connections between atoms. MACE‑H goes further by incorporating many‑body message passing: it can combine information from triplets and larger groups of atoms in a controlled way. A special node‑degree expansion module efficiently builds up higher‑order angular features without overwhelming memory and compute resources. This lets the model represent subtle patterns in the local chemical environment, such as the orientation of neighboring layers in twisted two‑dimensional materials or the complex bonding in bulk gold. At the same time, an additional edge‑update stage converts these rich atomic features into predictions for each block of the Hamiltonian matrix that links pairs of atomic orbitals.
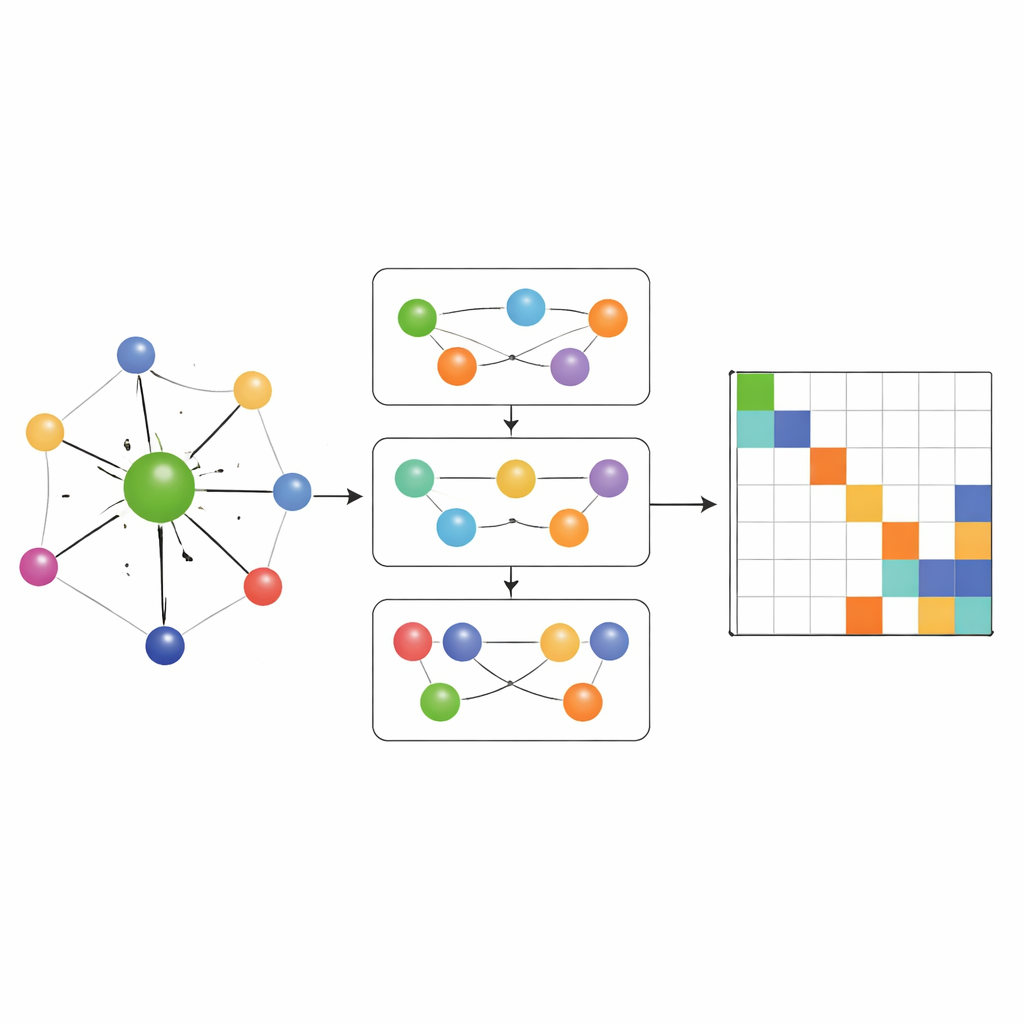
Accuracy, efficiency, and smart error checks
The researchers test MACE‑H on several demanding systems, including shifted and twisted bilayers of bismuth telluride and all‑electron calculations for bulk gold. Across these cases, the model predicts individual Hamiltonian matrix elements with average errors below a thousandth of an electron‑volt, and the resulting band structures and densities of states are visually indistinguishable from full quantum‑mechanical calculations. Compared with a strong previous model that uses only pairwise messages, MACE‑H is consistently more accurate and needs less training data to reach a given error level, while maintaining near‑linear scaling in system size. The architecture tends to focus strongly on the nearby atomic environment, which boosts data efficiency but slightly reduces sensitivity to very long‑range structural changes; however, even in those challenging twisted structures, the electronic properties near the Fermi level remain well reproduced. The authors also show that a carefully designed “shift‑and‑scale” step stabilizes training when different parts of the Hamiltonian vary over many orders of magnitude, and they propose using how well the predicted matrix obeys a basic symmetry (Hermiticity) as a fast, label‑free indicator of reliability.
Toward rapid materials discovery
In plain terms, MACE‑H learns to emulate a heavy quantum‑mechanical solver for electrons while keeping track of the full matrix structure that underlies key electronic properties. Because it is accurate, data‑efficient, and scalable, it can be plugged into existing electronic‑structure codes to accelerate band‑structure calculations, guide high‑throughput screening of candidate materials, or help simulations that couple electron motion to atomic motion. The approach is general enough to be extended to other quantum operators, such as density matrices, opening a route to faster self‑consistent calculations. As such models mature and gain robust uncertainty estimates, they are likely to become central tools in the virtual discovery and design of new electronic materials.
Citation: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Keywords: machine learning Hamiltonian, graph neural networks, electronic structure, materials discovery, density functional theory