Clear Sky Science · es
Predicción del hamiltoniano electrónico equivariante mediante paso de mensajes de muchos cuerpos
Por qué importa predecir los electrones más rápido
El diseño de nuevas baterías, chips y dispositivos cuánticos a menudo depende de comprender cómo se desplazan los electrones a través de un material. El estándar de oro para esto es un método cuántico llamado teoría del funcional de la densidad, que es preciso pero tremendamente lento para sistemas grandes o complejos. Este artículo presenta un nuevo modelo de aprendizaje automático, MACE‑H, que puede imitar estos costosos cálculos cuánticos para electrones con una exactitud notable, pero a una fracción del coste. Para los no especialistas, este trabajo apunta hacia cribados digitales más rápidos de materiales avanzados antes de que nadie corte metal o haga crecer un cristal.
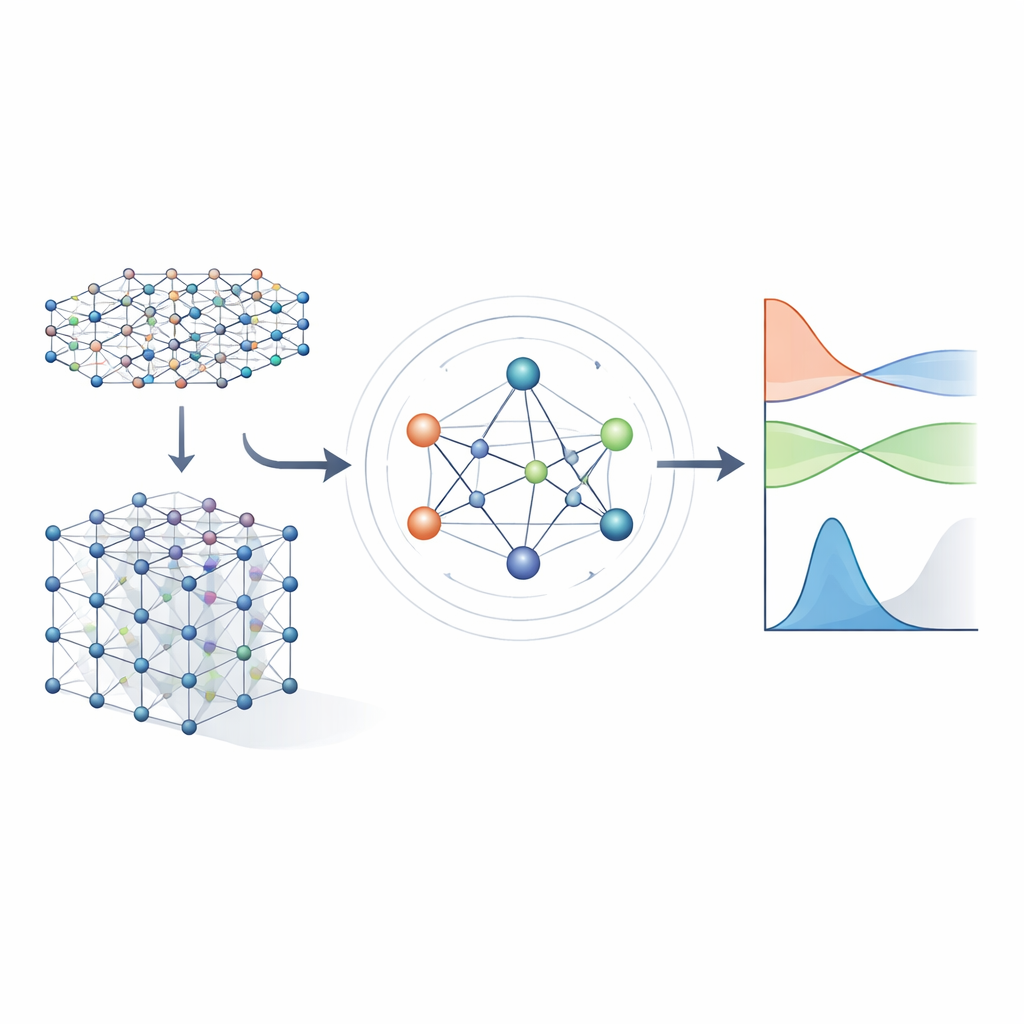
De ecuaciones pesadas a atajos aprendidos
Los métodos tradicionales de estructura electrónica funcionan resolviendo un gran objeto matemático llamado hamiltoniano, que codifica cómo interactúan los electrones con los núcleos atómicos y entre sí. Para materiales realistas, el hamiltoniano se representa como una matriz enorme cuya dimensión se dispara a medida que el sistema crece, haciendo que el cálculo directo sea cada vez más costoso. Enfoques anteriores de aprendizaje automático trataron de simplificar el problema ya sea usando modelos aproximados de tight‑binding o prediciendo únicamente propiedades escalares como energías. Estos enfoques pueden ser rápidos, pero por lo general no conservan suficiente detalle sobre la estructura electrónica para predecir de forma fiable bandas de energía, transporte u comportamiento óptico en una amplia variedad de materiales.
Una red neuronal que respeta la simetría
Los autores se basan en una nueva generación de redes neuronales que respetan explícitamente las simetrías del espacio tridimensional: rotaciones, reflexiones y traslaciones. En MACE‑H, los átomos de un material se tratan como nodos en un grafo, y el modelo transmite “mensajes” a lo largo de los enlaces entre ellos. Crucialmente, estos mensajes están diseñados de modo que si se rota o traslada todo el cristal, las características internas rotan y se mueven exactamente de la misma manera que dicta la física subyacente. Esto se consigue mediante una descomposición cuidadosa de la información en componentes que se transforman como vectores y objetos de orden superior bajo rotaciones. Al hacerlo, el modelo maneja de forma natural orbitales atómicos con diferentes formas, incluidas las más complejas que son importantes para elementos pesados y para efectos de acoplamiento espín‑órbita.
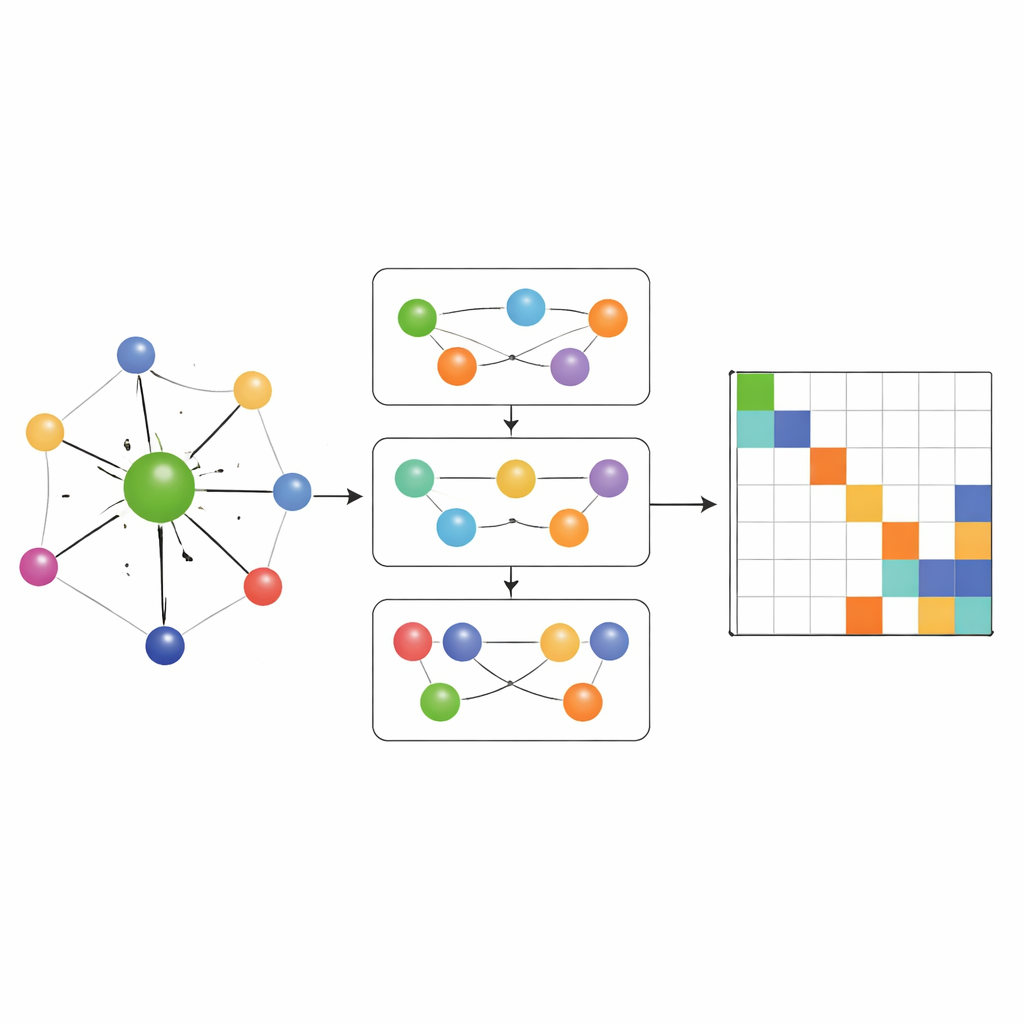
Capturar química de muchos cuerpos, no solo pares
La mayoría de las redes anteriores que aprenden hamiltonianos solo permitían que la información fluyera a través de conexiones pareadas simples entre átomos. MACE‑H va más allá al incorporar paso de mensajes de muchos cuerpos: puede combinar información de tripletes y grupos mayores de átomos de manera controlada. Un módulo especial de expansión por grado de nodo construye de forma eficiente características angulares de orden superior sin desbordar la memoria y los recursos de cómputo. Esto permite al modelo representar patrones sutiles en el entorno químico local, como la orientación de capas vecinas en materiales bidimensionales torcidos o el enlace complejo en oro macroscópico. Al mismo tiempo, una etapa adicional de actualización de aristas convierte estas ricas características atómicas en predicciones para cada bloque de la matriz hamiltoniana que enlaza pares de orbitales atómicos.
Exactitud, eficiencia y controles inteligentes de error
Los investigadores prueban MACE‑H en varios sistemas exigentes, incluidos bicapas desplazadas y torcidas de telururo de bismuto y cálculos de electrones completos para oro macroscópico. En estos casos, el modelo predice elementos individuales de la matriz hamiltoniana con errores medios por debajo de una milésima de electronvoltio, y las bandas de energía y densidades de estados resultantes son visualmente indistinguibles de los cálculos cuántico‑mecánicos completos. En comparación con un modelo previo potente que usa solo mensajes pareados, MACE‑H es sistemáticamente más preciso y necesita menos datos de entrenamiento para alcanzar un nivel de error dado, manteniendo al mismo tiempo una escalabilidad casi lineal con el tamaño del sistema. La arquitectura tiende a centrarse fuertemente en el entorno atómico cercano, lo que mejora la eficiencia de los datos pero reduce ligeramente la sensibilidad a cambios estructurales de muy largo alcance; sin embargo, incluso en esas estructuras torcidas desafiantes, las propiedades electrónicas cerca del nivel de Fermi se reproducen bien. Los autores también muestran que un paso cuidadosamente diseñado de “desplazar y escalar” estabiliza el entrenamiento cuando diferentes partes del hamiltoniano varían en muchas órdenes de magnitud, y proponen usar cuán bien la matriz predicha cumple una simetría básica (hermiticidad) como un indicador rápido y sin etiquetas de fiabilidad.
Hacia un descubrimiento rápido de materiales
En términos sencillos, MACE‑H aprende a emular un potente solucionador cuántico‑mecánico para electrones mientras mantiene la estructura matricial completa que subyace a propiedades electrónicas clave. Porque es preciso, eficiente en datos y escalable, puede integrarse en códigos de estructura electrónica existentes para acelerar cálculos de bandas, guiar cribados de alto rendimiento de materiales candidatos o ayudar simulaciones que acoplan el movimiento electrónico con el atómico. El enfoque es lo bastante general como para extenderse a otros operadores cuánticos, como matrices de densidad, abriendo una vía hacia cálculos auto‑consistentes más rápidos. A medida que estos modelos maduren y obtengan estimaciones de incertidumbre robustas, es probable que se conviertan en herramientas centrales en el descubrimiento y diseño virtual de nuevos materiales electrónicos.
Cita: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Palabras clave: hamiltoniano aprendizaje automático, redes neuronales de grafos, estructura electrónica, descubrimiento de materiales, teoría del funcional de la densidad