Clear Sky Science · sv
Ekvivariant förutsägelse av elektronisk Hamiltonoperator med många‑kropps meddelandeöverföring
Varför snabbare förutsägelse av elektroner spelar roll
Att utforma nya batterier, datorchip och kvantapparater hänger ofta på att förstå hur elektroner rör sig i ett material. Guldkopian för detta är en kvantmetod som kallas täthetsfunktionalteori, som är noggrann men plågsamt långsam för stora eller komplexa system. Denna artikel presenterar en ny maskininlärningsmodell, MACE‑H, som kan efterlikna dessa dyra kvantberäkningar för elektroner med anmärkningsvärd noggrannhet, men till en bråkdel av kostnaden. För icke‑specialister pekar arbetet mot snabbare digital screening av avancerade material innan någon skär i metall eller odlar en kristall.
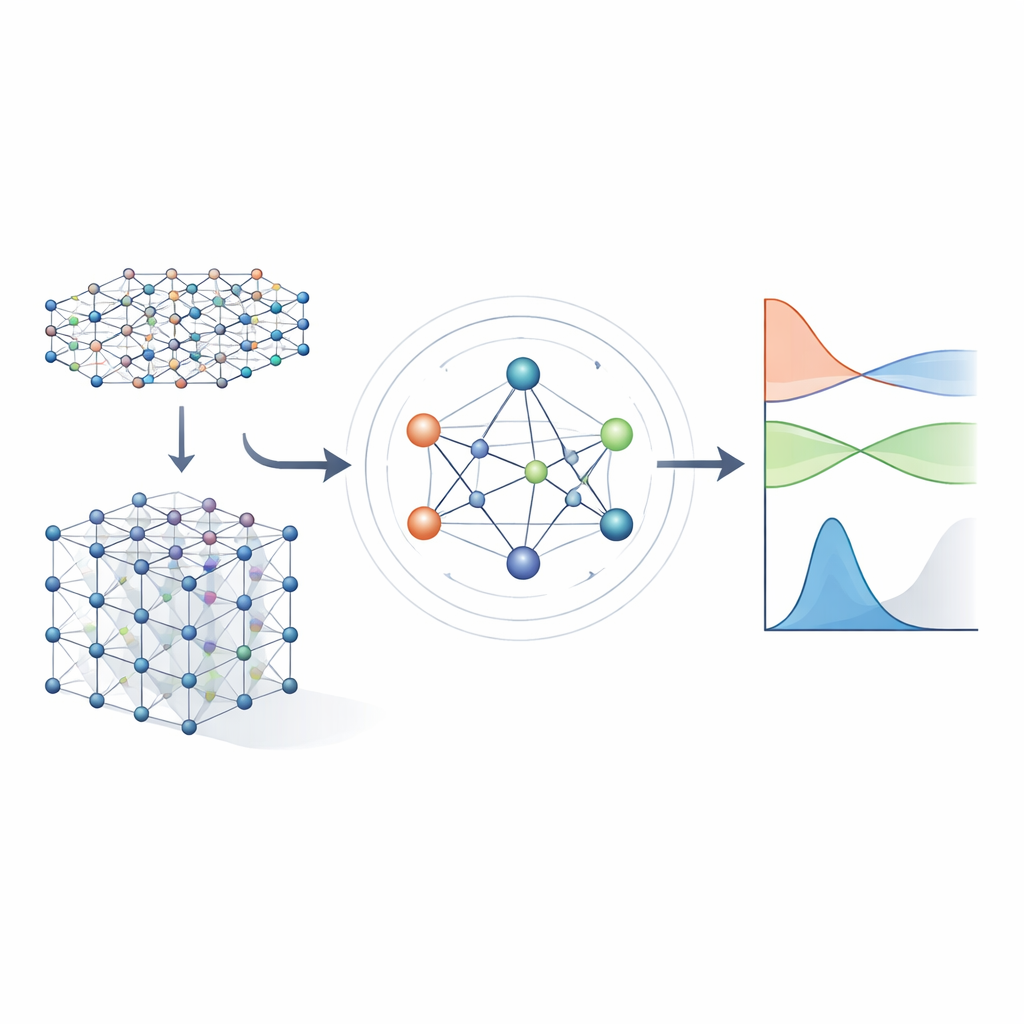
Från tunga ekvationer till inlärda genvägar
Traditionella metoder för elektronstruktur bygger på att lösa ett stort matematiskt objekt kallat Hamiltonoperatorn, som kodar hur elektroner interagerar med atomkärnor och med varandra. För realistiska material representeras Hamiltonoperatorn som en enorm matris vars storlek exploderar när systemet växer, vilket gör direkt beräkning allt dyrare. Tidigare maskininlärningstillvägagångssätt försökte förenkla problemet antingen genom approximativa tight‑binding‑modeller eller genom att förutsäga endast skalära egenskaper såsom energier. Dessa angreppssätt kan vara snabba, men behåller vanligtvis inte tillräckligt med detaljer om elektronstrukturen för att pålitligt förutsäga bandstrukturer, transport eller optiskt beteende över många olika material.
Neuralt nätverk som respekterar symmetri
Författarna bygger vidare på en ny generation av neurala nätverk som uttryckligen respekterar symmetrierna i tredimensionellt rum: rotationer, speglingar och translationer. I MACE‑H behandlas atomer i ett material som noder i en graf, och modellen skickar ”meddelanden” längs bindningarna mellan dem. Avgörande är att dessa meddelanden är utformade så att om man roterar eller flyttar hela kristallen, roterar och förflyttas de interna funktionerna på exakt samma sätt som den underliggande fysiken föreskriver. Detta uppnås genom en noggrann dekomposition av informationen i komponenter som transformeras som vektorer och högre ordningens objekt under rotation. Genom detta hanterar modellen naturligt atomorbitaler med olika former, inklusive de mer komplexa som är viktiga för tunga grundämnen och för spinn‑bana‑effekter.
Fånga många‑kropps‑kemi, inte bara par
De flesta tidigare nätverk som lärde Hamiltonoperatorer tillät endast informationsflöde längs enkla parvisa kopplingar mellan atomer. MACE‑H går längre genom att införliva många‑kropps meddelandeöverföring: den kan kombinera information från tripletter och större atomgrupper på ett kontrollerat sätt. En särskild modul för utvidgning efter nodgrad bygger effektivt upp högre‑ordnings vinkelaktiga funktioner utan att överväldiga minne och beräkningsresurser. Detta låter modellen representera subtila mönster i den lokala kemiska omgivningen, såsom orienteringen av intilliggande lager i tvistade tvådimensionella material eller den komplexa bindningen i massiv guld. Samtidigt omvandlar en ytterligare kantuppdateringsfas dessa rika atomära funktioner till förutsägelser för varje block i Hamiltonmatrisen som länkar par av atomorbitaler.
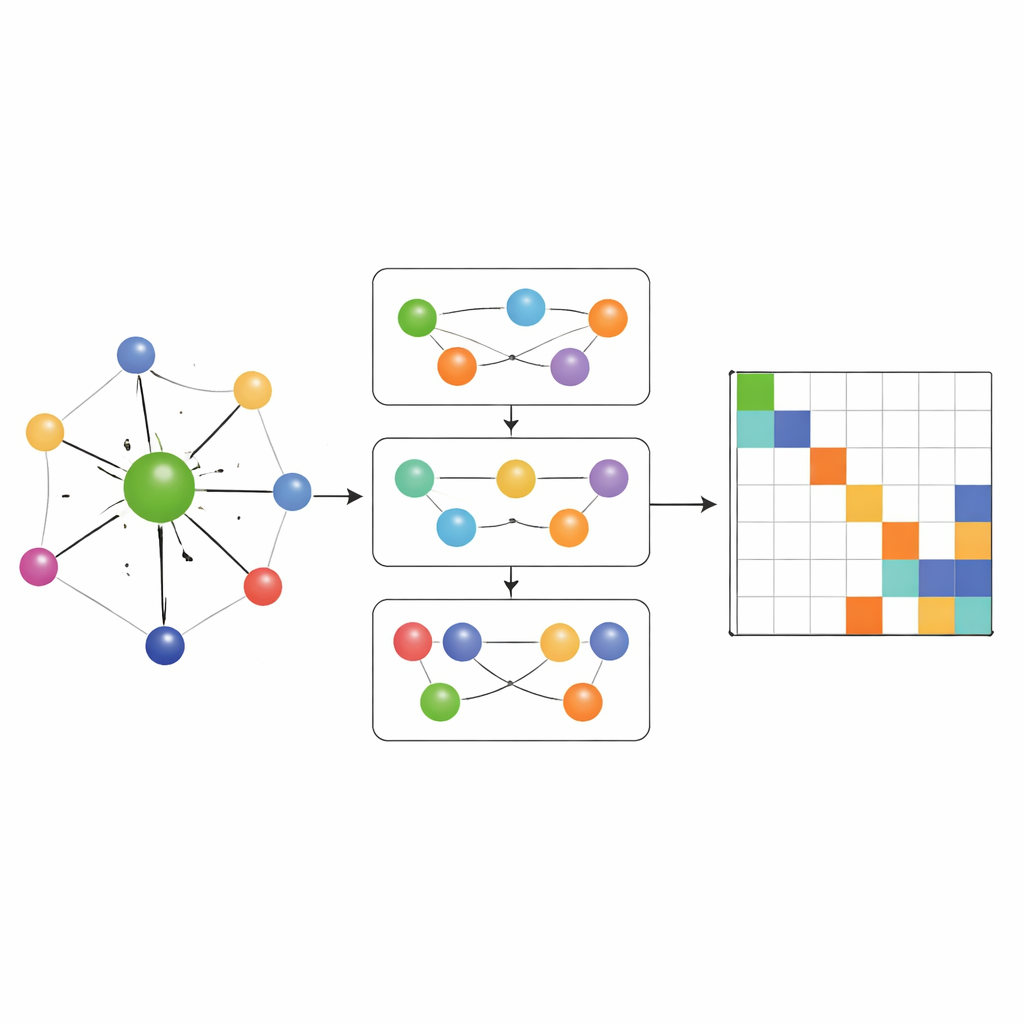
Noggrannhet, effektivitet och smarta felkontroller
Forskarna testar MACE‑H på flera krävande system, inklusive förskjutna och tvistade bilager av bismuttellurid och all‑electron‑beräkningar för massiv guld. I dessa fall förutspår modellen individuella Hamiltonmatriselement med genomsnittliga fel under en tusendels elektronvolt, och de resulterande bandstrukturerna och täthetsfunktionerna är visuellt omöjliga att skilja från fullständiga kvantmekaniska beräkningar. Jämfört med en stark tidigare modell som bara använder parvisa meddelanden är MACE‑H konsekvent mer noggrann och kräver mindre träningsdata för att nå en given felnivå, samtidigt som den behåller nära linjär skalning med systemstorlek. Arkitekturen tenderar att fokusera starkt på den närmaste atommiljön, vilket ökar dataeffektiviteten men något minskar känsligheten för mycket långräckta strukturella förändringar; även i de utmanande tvistade strukturerna återges dock de elektroniska egenskaperna nära Ferminivån väl. Författarna visar också att ett omsorgsfullt designat ”skifta‑och‑skala”‑steg stabiliserar träningen när olika delar av Hamiltonoperatorn varierar över många storleksordningar, och de föreslår att man använder hur väl den förutsagda matrisen uppfyller en grundläggande symmetri (Hermiticitet) som en snabb, etikettfri indikator på tillförlitlighet.
Mot snabb materialupptäckt
Med enkla ord lär sig MACE‑H att efterlikna en tung kvantmekanisk lösare för elektroner samtidigt som den beaktar hela matrisstrukturen som ligger bakom viktiga elektroniska egenskaper. Eftersom den är noggrann, dataeffektiv och skalbar kan den kopplas in i befintliga elektronstrukturkoder för att påskynda bandstruksberäkningar, styra höggenomströmning‑screening av kandidatmaterial eller hjälpa simuleringar som kopplar elektronrörelse till atomrörelse. Tillvägagångssättet är tillräckligt generellt för att kunna utvidgas till andra kvantoperatorer, såsom täthetsmatriser, vilket öppnar en väg till snabbare självkonsekventa beräkningar. När sådana modeller mognar och får robusta osäkerhetsuppskattningar är de sannolikt på väg att bli centrala verktyg i den virtuella upptäckten och designen av nya elektroniska material.
Citering: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Nyckelord: maskininlärning Hamiltonoperator, grafnätverk, elektronstruktur, materialupptäckt, täthetsfunktionalteori