Clear Sky Science · nl
Equivariantie elektronische Hamiltoniaan‑voorspelling met veel‑deeltjes berichtuitwisseling
Waarom het sneller voorspellen van elektronen ertoe doet
Het ontwerp van nieuwe batterijen, computerchips en kwantumapparaten hangt vaak af van inzicht in hoe elektronen zich door een materiaal bewegen. De gouden standaard hiervoor is een kwantummethode genaamd dichtheidsfunctionaaltheorie, die nauwkeurig maar pijnlijk traag is voor grote of complexe systemen. Dit artikel introduceert een nieuw machine‑learningmodel, MACE‑H, dat deze kostbare kwantumberekeningen voor elektronen kan nabootsen met opmerkelijke nauwkeurigheid, maar tegen een fractie van de kosten. Voor niet‑specialisten wijst dit werk op snellere digitale screening van geavanceerde materialen voordat er metaal wordt gesneden of een kristal wordt gegroeid.
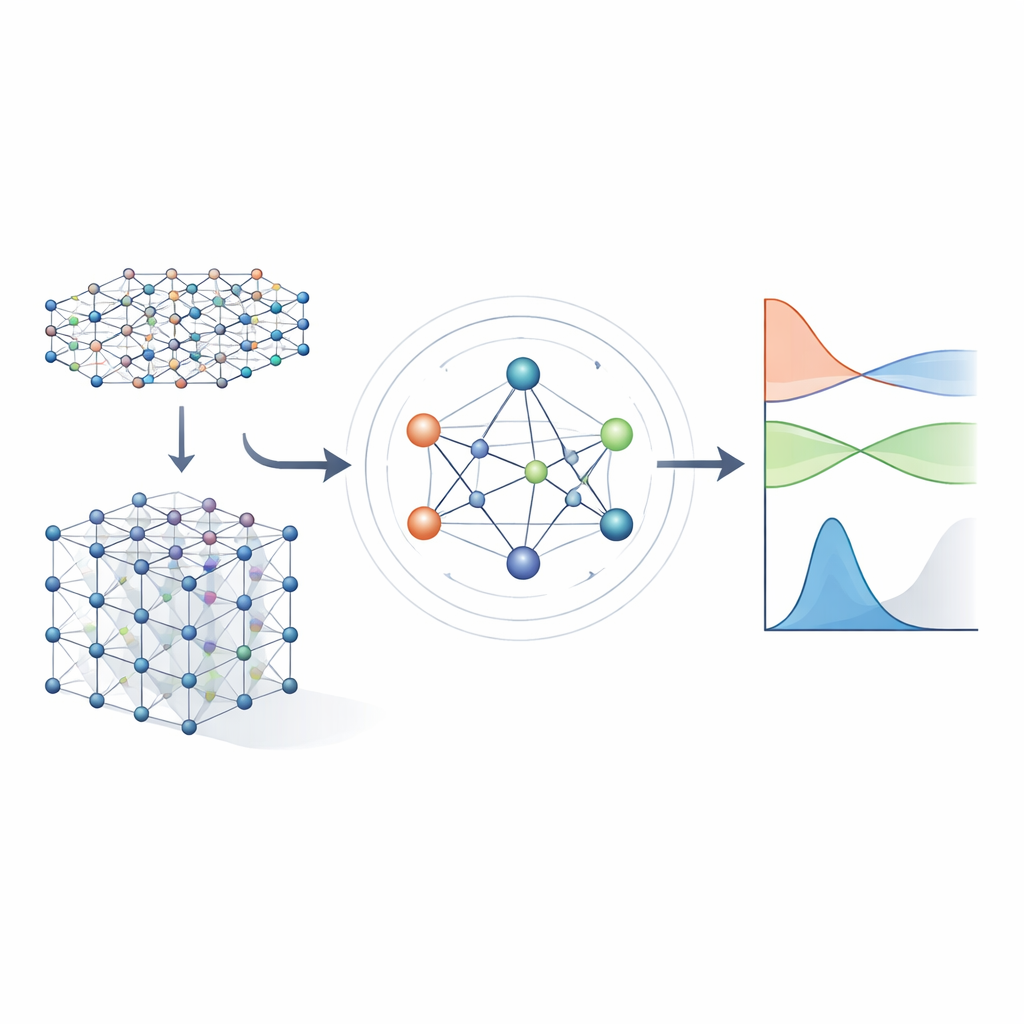
Van ingewikkelde vergelijkingen naar geleerde snelkoppelingen
Traditionele methoden voor elektronische structuur werken door het oplossen van een groot wiskundig object dat de Hamiltoniaan heet, en dat vastlegt hoe elektronen met atoomkernen en met elkaar interageren. Voor realistische materialen wordt de Hamiltoniaan weergegeven als een enorme matrix waarvan de omvang explodeert naarmate het systeem groeit, waardoor directe berekening steeds duurder wordt. Eerdere machine‑learningbenaderingen probeerden het probleem te vereenvoudigen door ofwel benaderende tight‑binding‑modellen te gebruiken of door alleen scalare eigenschappen zoals energieën te voorspellen. Deze benaderingen kunnen snel zijn, maar ze behouden meestal niet genoeg detail over de elektronische structuur om betrouwbaar bandstructuren, transport of optisch gedrag over veel verschillende materialen te voorspellen.
Een neuraal netwerk dat symmetrie respecteert
De auteurs bouwen voort op een nieuwe generatie neurale netwerken die expliciet de symmetrieën van de driedimensionale ruimte respecteren: rotaties, reflecties en translaties. In MACE‑H worden atomen in een materiaal behandeld als knopen in een graaf, en het model stuurt "berichten" langs de bindingen tussen hen. Cruciaal is dat deze berichten zo zijn ontworpen dat als je het hele kristal roteert of verplaatst, de interne kenmerken precies op dezelfde manier roteren en verschuiven als door de onderliggende fysica wordt voorgeschreven. Dit wordt bereikt door een zorgvuldige decompositie van de informatie in componenten die onder rotatie transformeren als vectoren en hogere‑orde objecten. Daarmee kan het model op natuurlijke wijze atomaire orbitalen met verschillende vormen verwerken, inclusief de complexere die belangrijk zijn voor zware elementen en voor spin‑baan‑effecten.
Veel‑deeltjeschemie vangen, niet alleen paren
De meeste eerdere Hamiltoniaan‑leer‑netwerken lieten informatie alleen stromen langs eenvoudige paargewijze verbindingen tussen atomen. MACE‑H gaat verder door veel‑deeltjes berichtuitwisseling te integreren: het kan informatie van tripletten en grotere groepen atomen op een gecontroleerde manier combineren. Een speciaal knoop‑graadexpansiemodule bouwt efficiënt hogere‑orde hoekfuncties op zonder het geheugen en de rekenmiddelen te overspoelen. Dit stelt het model in staat subtiele patronen in de lokale chemische omgeving weer te geven, zoals de oriëntatie van naburige lagen in gedraaide tweedimensionale materialen of de complexe bindingen in massief goud. Tegelijkertijd zet een extra rand‑updatefase deze rijke atomaire kenmerken om in voorspellingen voor elk blok van de Hamiltoniaan‑matrix die paren van atomaire orbitalen koppelt.
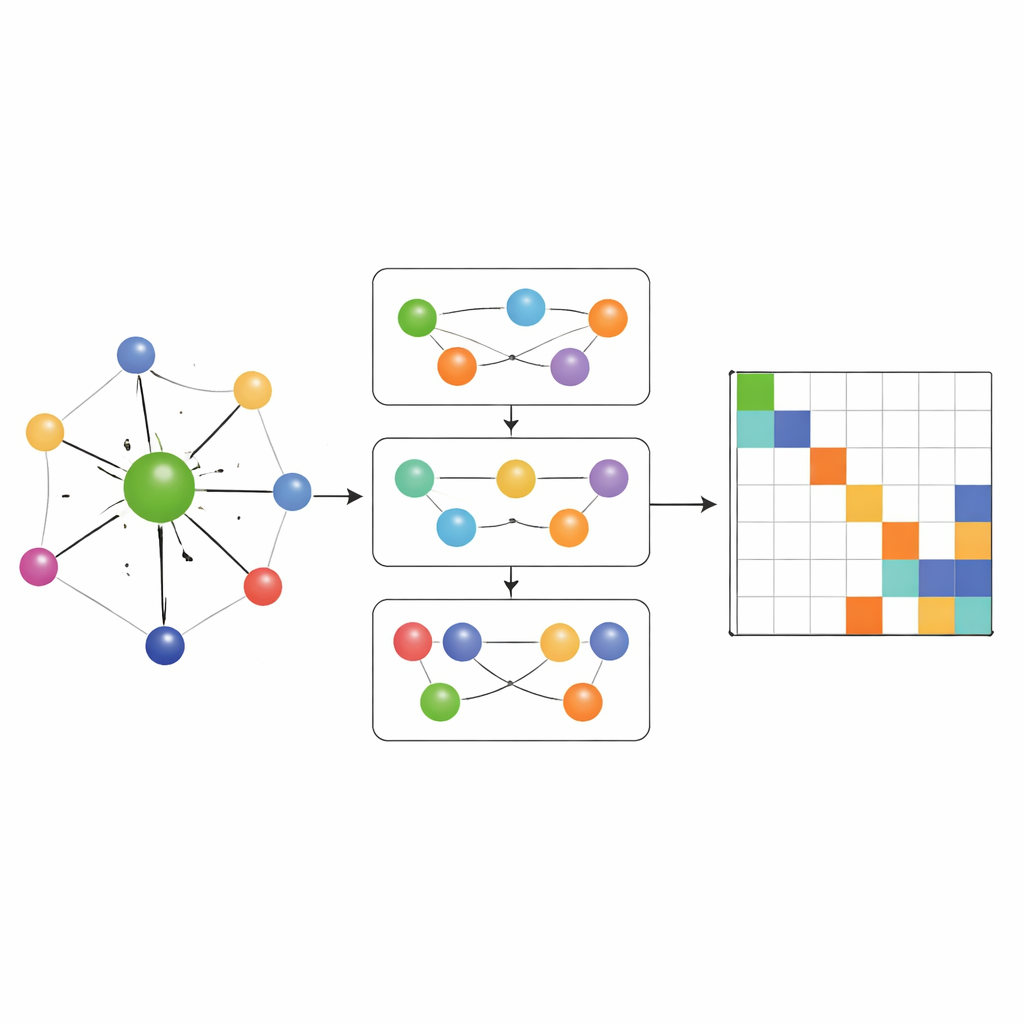
Nauwkeurigheid, efficiëntie en slimme foutcontroles
De onderzoekers testen MACE‑H op verschillende veeleisende systemen, waaronder verschoven en gedraaide bilagen van bismuttelluride en all‑electron berekeningen voor massief goud. In al deze gevallen voorspelt het model individuele Hamiltoniaan‑matrixelementen met gemiddelde fouten onder een duizendste van een elektronenvolt, en de resulterende bandstructuren en toestandsdichtheden zijn visueel niet te onderscheiden van volledige kwantummechanische berekeningen. Vergeleken met een sterk eerder model dat alleen paargewijze berichten gebruikt, is MACE‑H consequent nauwkeuriger en heeft het minder trainingsdata nodig om een gegeven foutniveau te bereiken, terwijl het bijna lineaire schaalbaarheid met systeemgrootte behoudt. De architectuur richt zich sterk op de nabije atomaire omgeving, wat de data‑efficiëntie verhoogt maar de gevoeligheid voor zeer langafstands structurele veranderingen iets vermindert; zelfs in die uitdagende gedraaide structuren blijven de elektronische eigenschappen nabij het Fermi‑niveau echter goed gereproduceerd. De auteurs tonen ook aan dat een zorgvuldig ontworpen "verschuif‑en‑schaal"‑stap het trainen stabiliseert wanneer verschillende delen van de Hamiltoniaan over vele ordes van grootte variëren, en ze stellen voor om hoe goed de voorspelde matrix een basis symmetrie (Hermitische eigenschap) naleeft te gebruiken als een snelle, label‑vrije indicator van betrouwbaarheid.
Op weg naar snelle materialenontdekking
In eenvoudige bewoordingen leert MACE‑H een zware kwantummechanische oplosser voor elektronen te emuleren terwijl het de volledige matrixstructuur bewaakt die ten grondslag ligt aan belangrijke elektronische eigenschappen. Omdat het nauwkeurig, data‑efficiënt en schaalbaar is, kan het worden ingebouwd in bestaande elektronische‑structuurcodes om bandstructuurberekeningen te versnellen, high‑throughput screening van kandidaatmaterialen te sturen, of simulaties te ondersteunen die elektronbeweging koppelen aan atoombeweging. De benadering is algemeen genoeg om te worden uitgebreid naar andere kwantumoperatoren, zoals dichtheidsmatrices, en opent daarmee een route naar snellere zelfconsistente berekeningen. Naarmate dergelijke modellen volwassen worden en robuuste onzekerheidsinschattingen krijgen, zullen ze waarschijnlijk centrale hulpmiddelen worden bij de virtuele ontdekking en het ontwerp van nieuwe elektronische materialen.
Bronvermelding: Qian, C., Vitartas, V., Kermode, J.R. et al. Equivariant electronic Hamiltonian prediction with many-body message passing. npj Comput Mater 12, 169 (2026). https://doi.org/10.1038/s41524-026-02020-1
Trefwoorden: machine learning Hamiltoniaan, graf neurale netwerken, elektronische structuur, materialenontdekking, dichtheidsfunctionaaltheorie