Clear Sky Science · tr
Kısıt-temelli orbital-optimalli uyarılmış durum yöntemi ile ΔSCF yöntemlerinin ayrıntılı bir karşılaştırması
Işık ve moleküller için neden önemli
Moleküller ışık absorbe ettiğinde, elektronları daha yüksek enerjili “uyarılmış” durumlara itilir. Bu uyarılmaları doğru şekilde tahmin etmek güneş pilleri, LED’ler, fotokatalizörler ve hatta radyasyonun biyolojik doku üzerindeki etkisini anlamak için esastır. Bu makale, kimyagerlerin uyarılmış elektronları simüle etmek için kullandığı iki bilgisayar yöntemi ailesinin perde arkasını inceliyor ve daha yeni bir yaklaşımın bu hesaplamaları hem daha güvenilir hem de daha çok yönlü hale getirebileceğini gösteriyor.
Uyarılmış elektronları genellikle nasıl simüle ediyoruz
Işık–madde etkileşimlerinin çoğu modern simülasyonu, elektronların küçük rahatsızlıklara nasıl yanıt verdiğini ele alan bir iş atı çerçevesi olan zaman-bağımlı yoğunluk fonksiyonel teorisini (TD-DFT) kullanır. Makul derecede doğru ve bilgisayarda çalıştırması nispeten ucuz olduğu için popülerdir. Ancak bu standart yaklaşım en önemli durumların bazılarıyla zorlanır: yükün uzun mesafelere taşındığı hallerde, iç (çekirdek) elektronların uyarıldığı durumlarda veya birden fazla elektron aynı anda uyarıldığında. Bu kör noktaların etrafından dolaşmak için kimyagerler giderek orbital-optimize edilmiş yöntemlere yöneliyor. Burada uyarılmış bir durum doğrudan kurulur ve yerel durumun bir tepkisi olarak çıkarılmak yerine öz-uyumlu bir prosedürle rafine edilir.
Eski araçlar: elektronları yeni yuvalara zorlamak
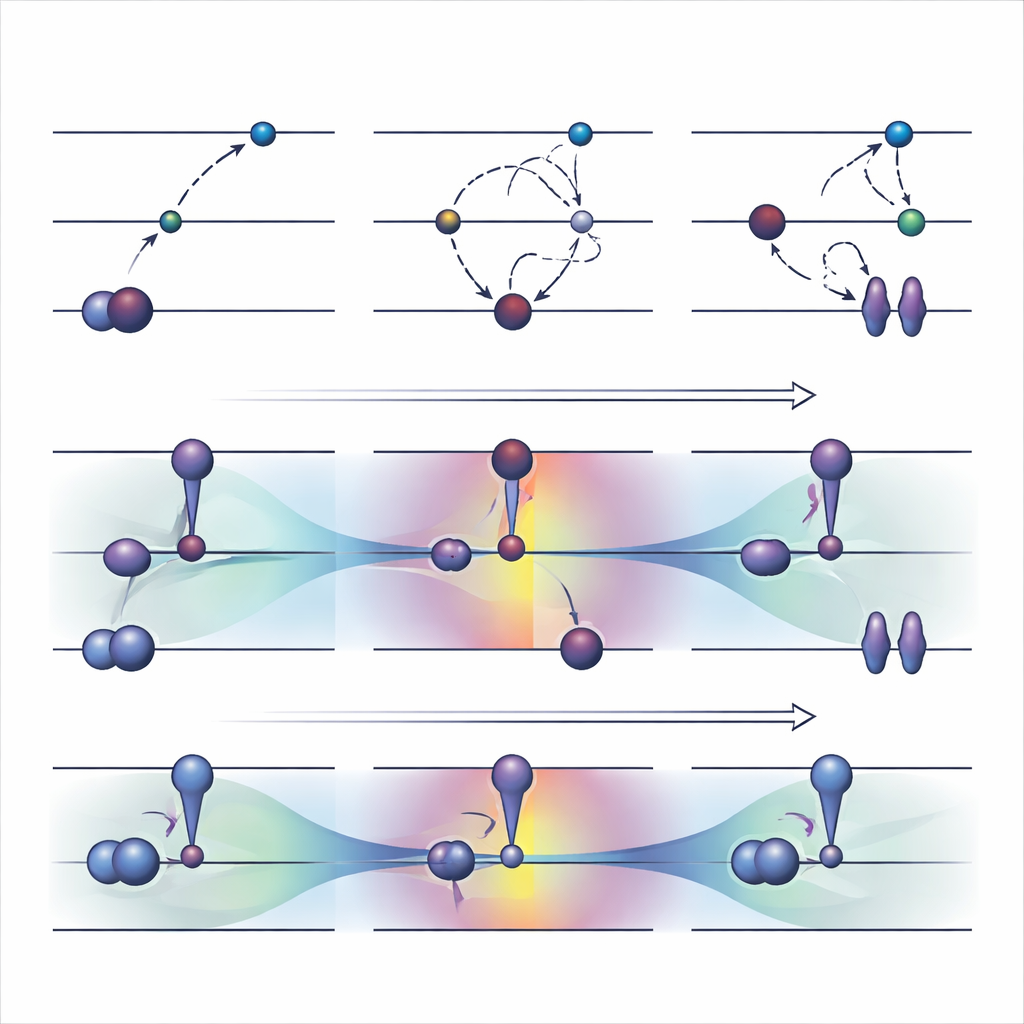
Yaygın kullanılan bir orbital-optimize strateji olan ΔSCF, elektronlar için alışılmış doldurma kuralını açıkça ihlal ederek çalışır: bir dolu orbital boşaltılır ve daha yüksek bir orbital doldurulur, ardından sistem bu yeni düzene göre yeniden optimize edilir. Temel ΔSCF’nin üzerinde, maksimum örtüşme yöntemi gibi pratik algoritmalar hesabın temel duruma geri kaymasını engellemeye çalışır. Bu yaklaşımlar zor uyarılmaları tanımlayabilir ve maliyeti normal bir temel durum çalışmasıyla yaklaşık olarak aynıdır. Ancak ciddi dezavantajları vardır. Genellikle yavaş yakınsar veya hiç yakınsamaz, sessizce yanlış duruma geri dönebilir ve büyük ölçüde iki açıkça tanımlanmış orbital arasında tek-elektron sıçraması gibi gözüken uyarılmalarla sınırlıdır.
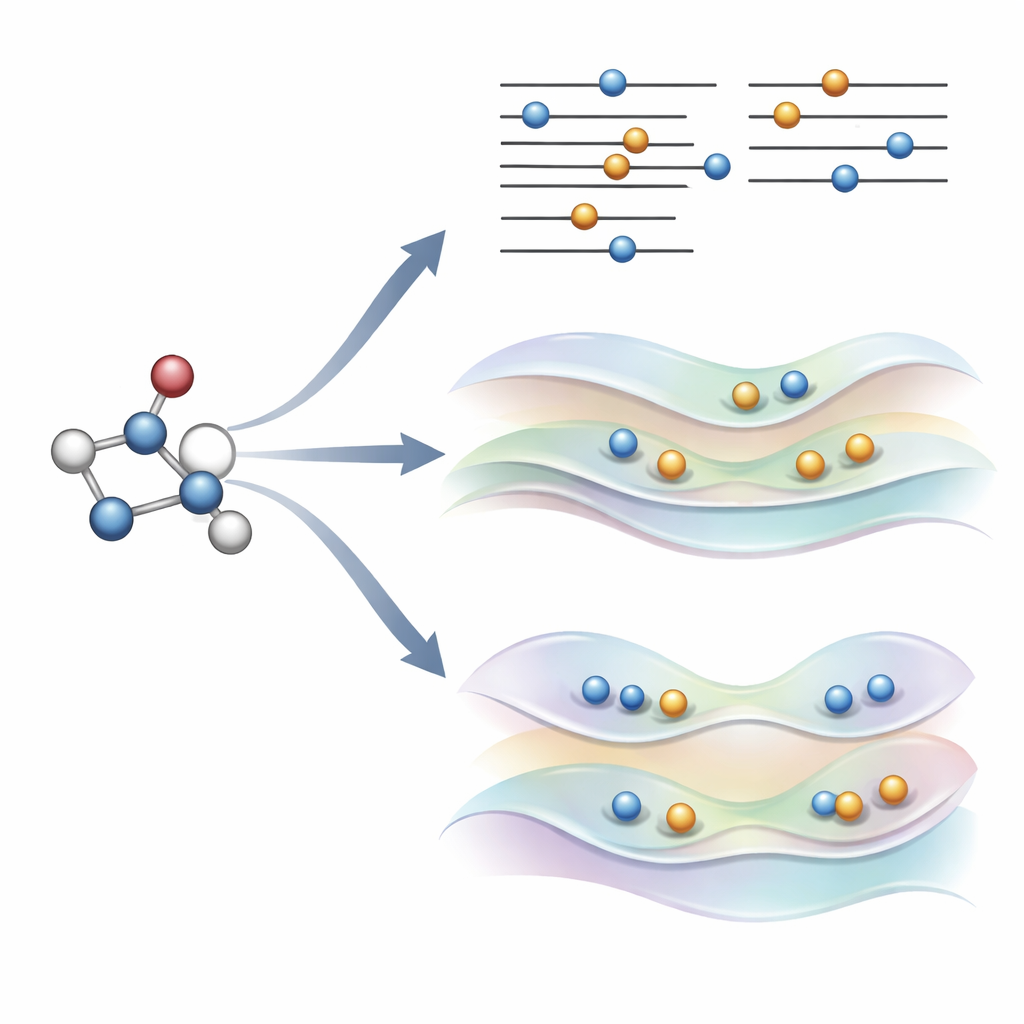
Yeni fikir: kuralları çiğnemek yerine orbitalere nazikçe yön vermek
Yazarlar yakın zamanda, alışılmış elektron doldurma kurallarını koruyan ve bunun yerine orbitaleri kendilerini uyarılmış durum şekline iten dikkatle tasarlanmış bir kısıt ekleyen alternatif bir yöntem olan COOX’u önerdiler. Bu çalışmada, iki yaklaşımın doğrudan karşılaştırılabilmesi için ΔSCF’deki aynı basit orbital sıçramalarını taklit etmek üzere inşa edilmiş bir sürüm olan ΔCOOX’a odaklanıyorlar. İşgörevleri elle değiştirmek yerine, ΔCOOX belirli orbitallerin enerjisini seçici olarak yükseltip düşüren ek bir potansiyel ekler; böylece istenen elektron etkin bir şekilde taşınmış olur. Bu, kısıtlı yoğunluk fonksiyonel teorisinin esnek çerçevesi içinde yapılır ve mevcut simülasyon kodlarına yalnızca mütevazı bir değişiklik gerektirir.
Çeşitli uyarılmalar üzerinde yan yana testler
Bu yöntemlerin pratikte nasıl performans gösterdiğini değerlendirmek için ekip, geniş bir moleküler uyarılma yelpazesinde kapsamlı testler yürüttü. Benzenin düşük-enerjili uyarılmış durumları için ΔCOOX tutarlı şekilde yaklaşık on adımdan az sürede bir çözüme ulaştı, oysa ΔSCF tabanlı teknikler bazen onlarca adım gerektirdi veya tamamen başarısız oldu. Ancak ΔSCF nihaî olarak amaçlanan duruma yakınsadığında, öngörülen uyarılma enerjileri genellikle ΔCOOX’unkilerle benzerdi. Atomlar ve küçük moleküller üzerinde yapılan sistematik karşılaştırmalar, her iki yaklaşımın sıradan değer uyarılmaları, Rydberg durumları ve iki elektronun terfi ettiği gerçek çift uyarılmalar için deneysel değerlerle iyi eşleşebildiğini gösterdi. Bununla birlikte ΔCOOX çok daha sağlam çıktı: nadiren yanlış elektronik desenine yakınsadığı görüldü ve ağır atomlar, uzak fragmanlar arasında güçlü yük aktarımı veya karmaşık ortamlardaki moleküller gibi zorlu durumlarda bile kararlı kaldı.
Zorlu durumlara ve karmaşık çevrelere ulaşma
ΔCOOX, istenen uyarılmayı kısıtı aracılığıyla zorunlu kıldığı için elektronik desenin hedefle yapısal olarak eşleşmesini garanti eder. Bu, bir hesabın sapıp sapmadığını fark etmeyi ve aynı anda birkaç orbitali içeren karışık uyarılmaları keşfetmeyi kolaylaştırır. Yazarlar ayrıca COOX’un, bir dolu kafes içindeki küçük bir misafir molekülü veya protein-benzeri bir ortamdaki kromofor gibi daha büyük sistemlere gömülü molekülleri doğal şekilde ele alacak biçimde genişletilebileceğini gösteriyor. Bu durumlarda uyarılmış durum önce ilgili molekül üzerinde tanımlanır ve ardından çevrenin tam kuantum tanımına sorunsuz şekilde gömülür; ΔSCF’nin sıkça gerektirdiği zahmetli elle orbital tutanak tutma işinden kaçınılır.
Gelecek simülasyonlar için anlamı
Çalışma, kısıt-temelli ΔCOOX yönteminin yerleşik ΔSCF şemaları kadar en az doğru olduğunu ve sayısal kararlılık ve güvenilirlik açısından genellikle açıkça üstün olduğunu sonucuna varıyor. Orbitaller arasında hareket eden tek bir elektronla tanımlanabilen çoğu uyarılmış durum için ΔCOOX, varsayılan tercih aracı olarak güçlü bir aday olarak ortaya çıkıyor ve ayrıca eski ΔSCF algoritmalarının yakınsamasına yardımcı olacak akıllı bir başlangıç noktası görevi görebilir. Daha geniş anlamda, tam COOX çerçevesi karmaşık uyarılmaları ve gerçekçi kimyasal ortamları rutin olarak ele almaya kapı açıyor; böylece uyarılmış durum simülasyonlarını gerçek dünya fotokimya ve malzeme bilimi alanlarındaki dağınık, çeşitli sistemlere daha da yaklaştırıyor.
Atıf: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Anahtar kelimeler: uyarılmış durumlar, yoğunluk fonksiyonel teorisi, elektronik yapı, yük aktarımı, hesaplamalı kimya