Clear Sky Science · sv
En detaljerad jämförelse av ΔSCF-metoder med den restriktionsbaserade orbitaloptimerade exciterade tillståndsmetoden
Varför detta spelar roll för ljus och molekyler
När molekyler absorberar ljus skjuts deras elektroner upp i högre energinivåer, så kallade "exciterade" tillstånd. Att korrekt förutsäga dessa excitationer är avgörande för att förstå solceller, lysdioder, fotokatalysatorer och till och med hur strålning påverkar biologisk vävnad. Denna artikel granskar två familjer av datorbaserade metoder som kemister använder för att simulera exciterade elektroner och visar hur ett nyare tillvägagångssätt kan göra dessa beräkningar både mer tillförlitliga och mer flexibla.
Hur vi vanligtvis simulerar exciterade elektroner
De flesta moderna simuleringar av ljus–materia-interaktioner använder en arbetsbockram som kallas tidsberoende täthetsfunktionalteori, vilken behandlar hur elektroner svarar på små störningar. Den är populär eftersom den är relativt exakt och förhållandevis billig att köra på en dator. Men detta standardtillvägagångssätt har svårigheter med några av de viktigaste fallen: när laddning flyttas över långa avstånd, när inre (kärn)elektroner exciteras, eller när mer än en elektron exciteras samtidigt. För att kringgå dessa blindfläckar har kemister i allt större utsträckning vänt sig till så kallade orbitaloptimerade metoder. Här byggs ett exciterat tillstånd upp direkt och förfinas sedan genom en självkonsekvent procedur, istället för att härledas som ett svar från grundtillståndet.
Gamla verktyg: tvinga elektroner till nya platser
En allmänt använd orbitaloptimerad strategi, kallad ΔSCF, fungerar genom att explicit bryta mot den vanliga fyllnadsregeln för elektroner: den tömmer en ockuperad orbital och fyller en högre, för att sedan reoptimera systemet kring detta nya mönster. Utöver grundläggande ΔSCF försöker praktiska algoritmer som maximum overlap method hindra att beräkningen glider tillbaka till grundtillståndet. Dessa tillvägagångssätt kan beskriva svåra excitationer och kostar ungefär lika mycket som en vanlig grundtillståndsberäkning. De har dock allvarliga nackdelar. De konvergerar ofta långsamt eller inte alls, kan tyst återgå till fel tillstånd och är i stor utsträckning begränsade till excitationer som ser ut som ett enkelt en-elektronshopp mellan två tydligt identifierade orbitaler.
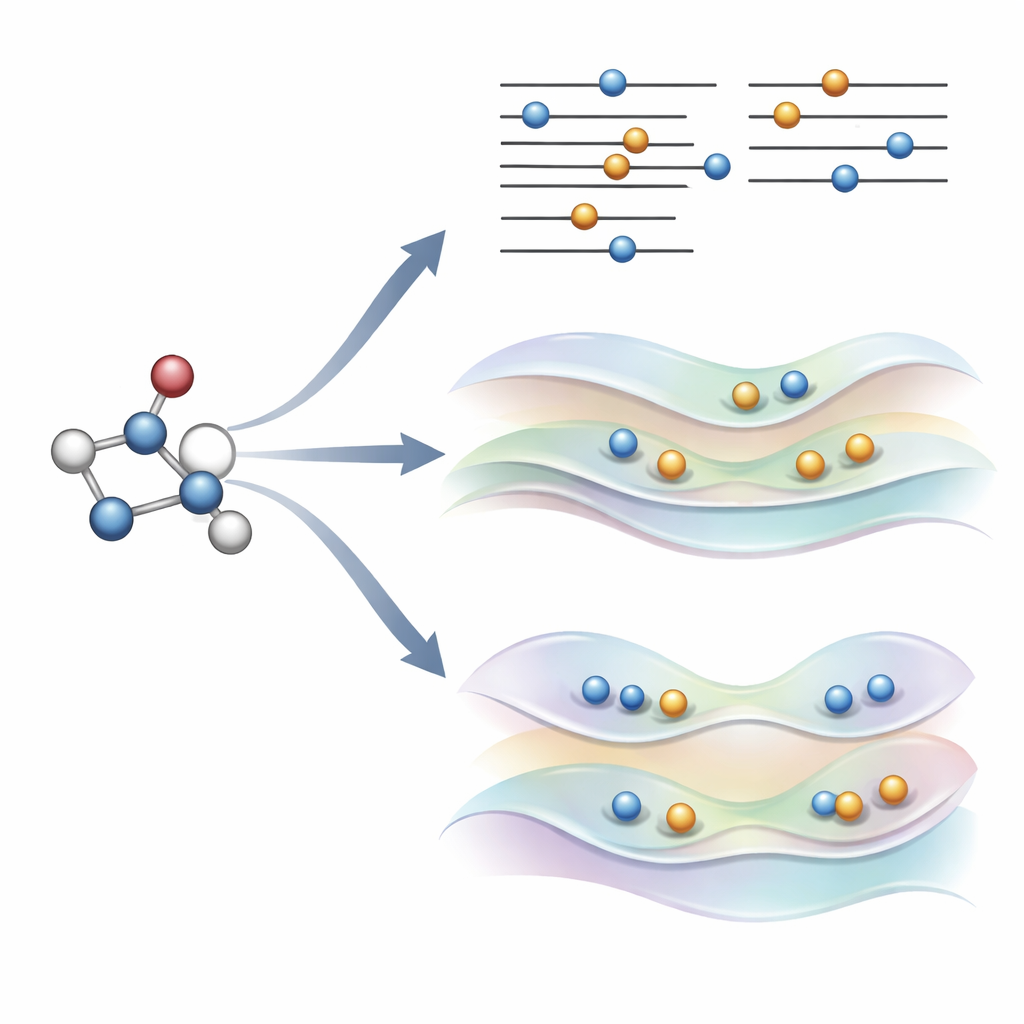
En ny idé: styra orbitalerna försiktigt istället för att bryta regler
Författarna föreslog nyligen ett alternativ, kallat COOX, som behåller de vanliga elektronfyllnadsreglerna intakta och istället lägger till en noggrant utformad restriktion för att föra orbitalerna själva mot en exciterad form. I denna studie fokuserar de på en version benämnd ΔCOOX, som är utformad för att efterlikna samma enkla orbitalhopp som används i ΔSCF så att de två tillvägagångssätten kan jämföras direkt. Istället för att manuellt ändra ockupationer lägger ΔCOOX till en extra potential som selektivt höjer och sänker energin hos specifika orbitaler tills den önskade elektronen effektivt har flyttats. Detta görs inom den flexibla ramen för begränsad täthetsfunktionalteori och kräver endast en måttlig modifiering av befintliga simulationskoder.
Sida vid sida-tester på många typer av excitationer
För att bedöma hur dessa metoder fungerar i praktiken körde teamet omfattande tester på ett brett spektrum av molekylära excitationer. För lågt liggande exciterade tillstånd i bensen nådde ΔCOOX konsekvent en lösning på färre än cirka tio steg, medan ΔSCF-baserade tekniker ibland behövde många tiotals steg eller misslyckades helt. Men när ΔSCF väl konvergerade till det avsedda tillståndet var de förutsagda excitationsenergierna i allmänhet liknande dem från ΔCOOX. Systematiska jämförelser på atomer och små molekyler visade att båda metoderna kan matcha experimentella värden väl för vanliga valensexcitationer, Rydberg-tillstånd och verkliga dubbelexcitationer där två elektroner promoveras. Däremot visade sig ΔCOOX vara mycket mer robust: den konvergerade sällan till fel elektroniskt mönster och förblev stabil även för utmanande fall med tunga atomer, stark laddningsöverföring mellan avlägsna fragment eller molekyler i komplexa omgivningar.
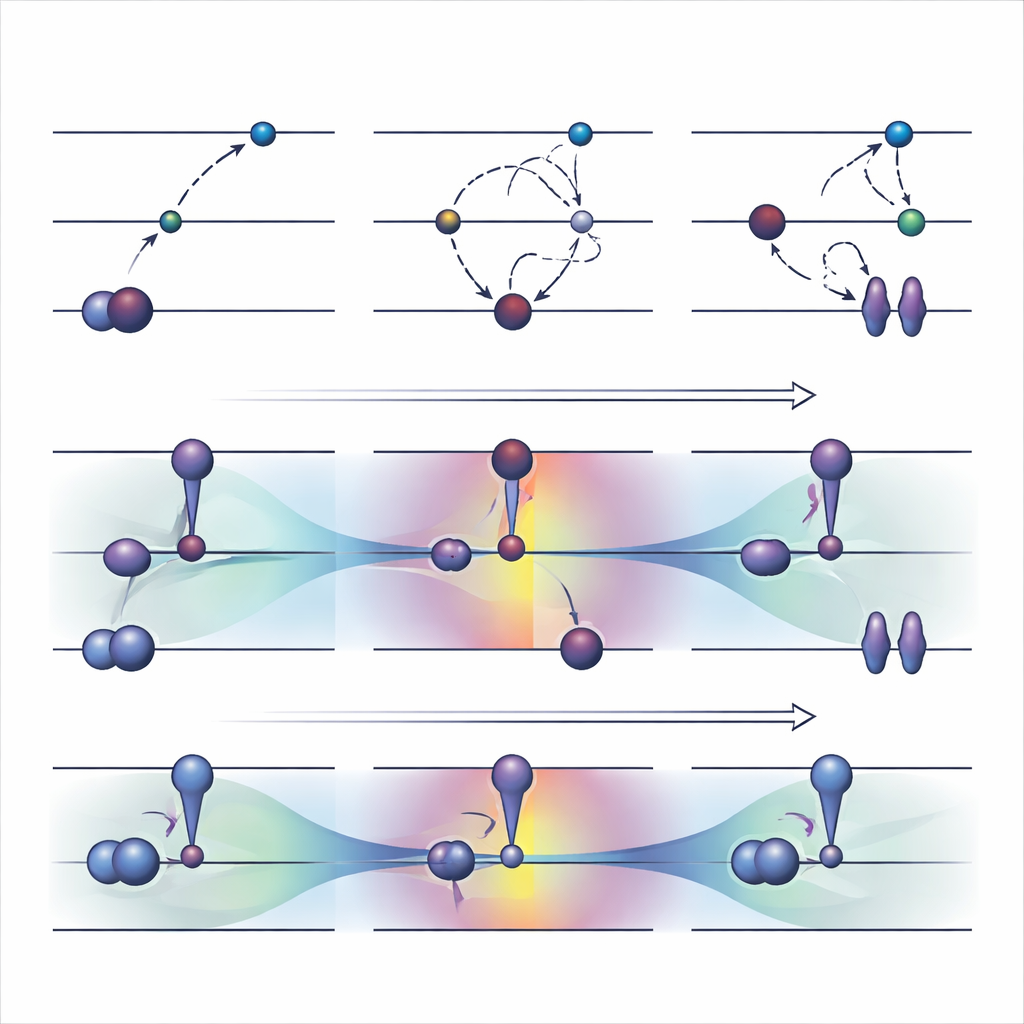
Nå svåra tillstånd och komplexa omgivningar
Eftersom ΔCOOX säkerställer den önskade excitationen genom sin restriktion garanterar den att det elektroniska mönstret stämmer överens med målet per konstruktion. Det gör det enklare att upptäcka när en beräkning har spårat ur och att utforska blandade excitationer som involverar flera orbitaler samtidigt. Författarna visar också att COOX naturligt kan utvidgas för att behandla molekyler inbäddade i större system, såsom en liten gäst inuti en fulleren-kapsel eller ett kromofor i en proteinliknande omgivning. I dessa fall definieras det exciterade tillståndet först på den molekyl som är av intresse och bäddas sedan sömlöst in i den fullständiga kvantmekaniska beskrivningen av omgivningen, vilket undviker det tidsödande manuella orbitalbokförandet som ofta krävs vid ΔSCF.
Vad detta innebär för framtida simuleringar
Studien slutar med slutsatsen att den restriktionsbaserade ΔCOOX-metoden är åtminstone lika exakt som etablerade ΔSCF-scheman och ofta klart överlägsen vad gäller numerisk stabilitet och tillförlitlighet. För de flesta exciterade tillstånd som kan beskrivas som en enkel elektron som rör sig mellan orbitaler framstår ΔCOOX som en stark kandidat för standardverktyget, och den kan också fungera som en smart utgångspunkt som hjälper äldre ΔSCF-algoritmer att konvergera. Mer allmänt öppnar hela COOX-ramverket dörren för att rutinmässigt behandla komplicerade excitationer och realistiska kemiska miljöer, och förflyttar simuleringar av exciterade tillstånd närmare de röriga, mångsidiga systemen som förekommer i verklig fotokemi och materialvetenskap.
Citering: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Nyckelord: exciterade tillstånd, täthetsfunktionalteori, elektronstruktur, laddningsöverföring, beräkningskem