Clear Sky Science · de
Ein detaillierter Vergleich von ΔSCF-Methoden mit der restriktionsbasierten orbitaloptimierten Zustandsmethode
Warum das für Licht und Moleküle wichtig ist
Wenn Moleküle Licht absorbieren, werden ihre Elektronen in energiereichere „angeregte“ Zustände gehoben. Diese Anregungen genau vorherzusagen ist unerlässlich, um Solarzellen, LEDs, Photokatalysatoren und sogar die Wirkungen von Strahlung auf biologisches Gewebe zu verstehen. Diese Arbeit blickt unter die Haube zweier Familien von Rechenmethoden, die Chemiker zur Simulation angeregter Elektronen verwenden, und zeigt, wie ein neuerer Ansatz diese Berechnungen sowohl verlässlicher als auch vielseitiger machen kann.
Wie wir angeregte Elektronen üblicherweise simulieren
Die meisten modernen Simulationen von Licht–Materie-Wechselwirkungen verwenden ein etabliertes Rahmenwerk, die zeitabhängige Dichtefunktionaltheorie, die beschreibt, wie Elektronen auf kleine Störungen reagieren. Sie ist beliebt, weil sie hinreichend akkurat und relativ kostengünstig auf einem Rechner ist. Dieser Standardansatz hat jedoch Probleme bei einigen der wichtigsten Fälle: wenn Ladung über große Entfernungen verschoben wird, wenn innere (Core-)Elektronen angeregt werden oder wenn mehr als ein Elektron gleichzeitig angeregt ist. Um diese blinden Flecken zu umgehen, greifen Chemiker zunehmend zu sogenannten orbitaloptimierten Methoden. Hier wird ein angeregter Zustand direkt konstruiert und dann durch ein selbstkonsistentes Verfahren verfeinert, anstatt ihn als Antwort des Grundzustands abzuleiten.
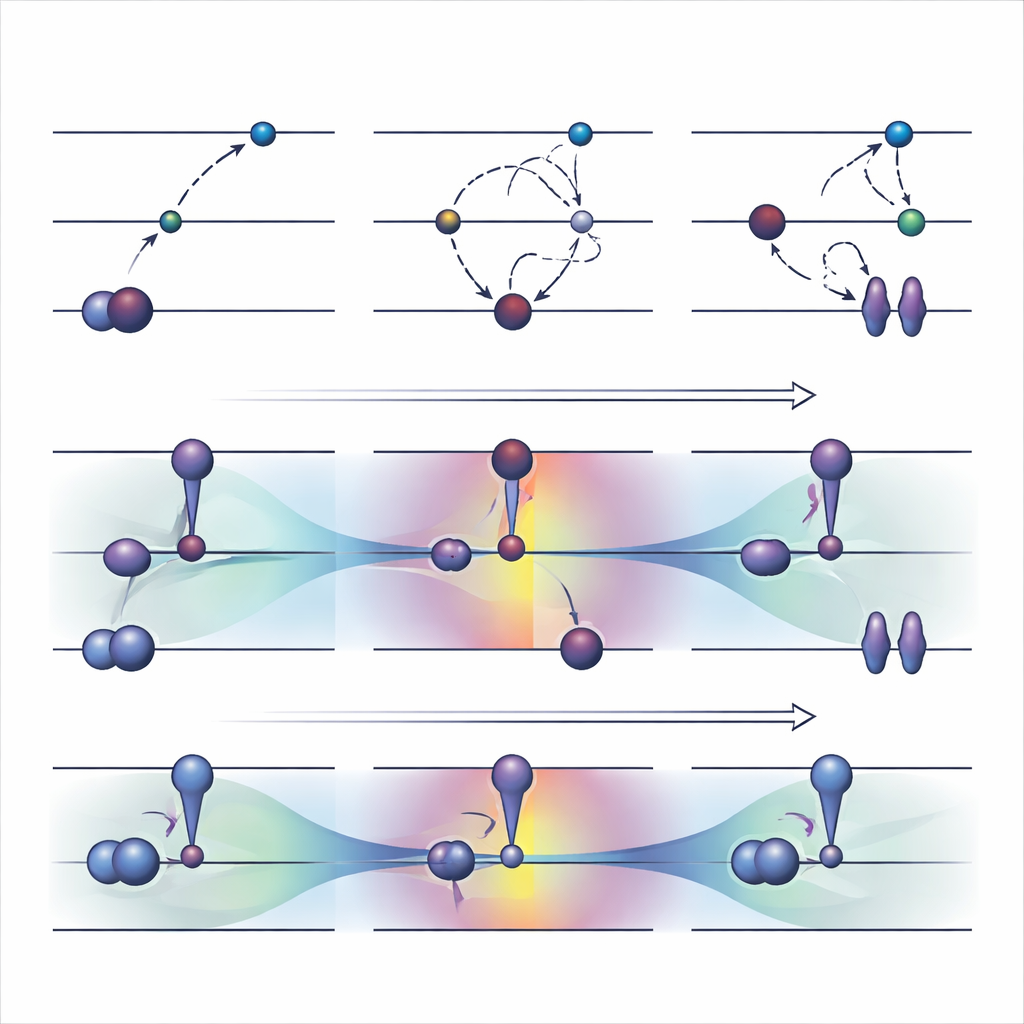
Alte Werkzeuge: Elektronen gewaltsam in neue Plätze schieben
Eine weit verbreitete orbitaloptimierte Strategie, genannt ΔSCF, arbeitet, indem sie die übliche Auffüllregel für Elektronen explizit verletzt: Ein besetztes Orbital wird entleert und ein höheres gefüllt, dann wird das System um dieses neue Besetzungsmuster herum reoptimiert. Auf die einfache ΔSCF-Methode aufbauende praktische Algorithmen wie die Maximum-Overlap-Methode versuchen zu verhindern, dass die Rechnung wieder in den Grundzustand zurückrutscht. Diese Ansätze können schwierige Anregungen beschreiben und kosten ungefähr so viel Rechenaufwand wie ein normaler Grundzustandslauf. Sie haben jedoch ernsthafte Nachteile. Sie konvergieren oft langsam oder gar nicht, können sich stillschweigend in den falschen Zustand zurückfallen lassen und sind weitgehend auf Anregungen beschränkt, die wie ein einfacher Ein-Elektron-Sprung zwischen zwei klar identifizierten Orbitalen aussehen.
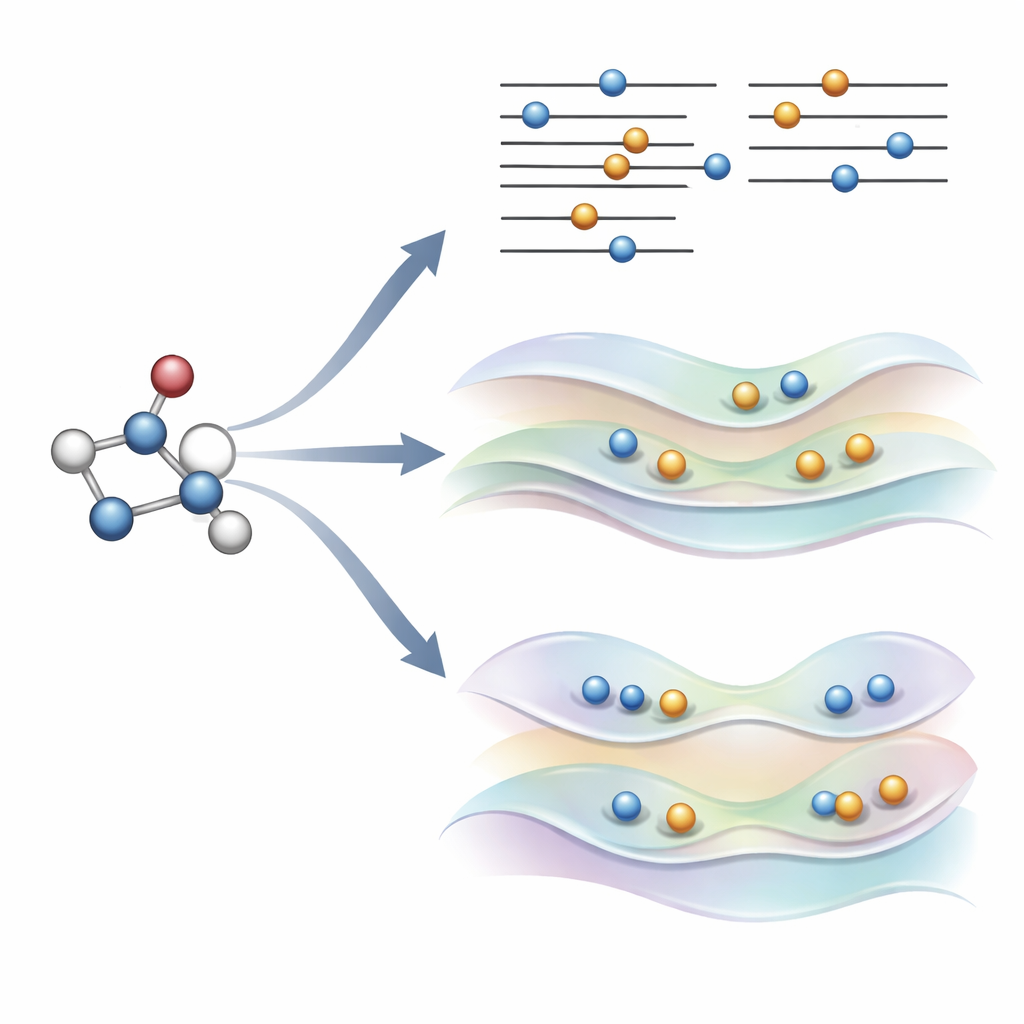
Eine neue Idee: Orbitale sanft steuern statt Regeln zu brechen
Die Autoren schlugen kürzlich eine Alternative vor, genannt COOX, die die üblichen Auffüllregeln beibehält und stattdessen eine sorgfältig gestaltete Einschränkung hinzufügt, um die Orbitale selbst in eine angeregten Zustandform zu drängen. In dieser Studie konzentrieren sie sich auf eine Variante mit der Bezeichnung ΔCOOX, die so konzipiert ist, dass sie die gleichen einfachen Orbitalübergänge nachahmt, die in ΔSCF verwendet werden, damit die beiden Ansätze direkt verglichen werden können. Anstatt Besetzungen manuell zu ändern, fügt ΔCOOX ein zusätzliches Potential hinzu, das selektiv die Energie bestimmter Orbitale anhebt oder senkt, bis das gewünschte Elektron effektiv verschoben ist. Dies geschieht im flexiblen Rahmen der eingeschränkten Dichtefunktionaltheorie und erfordert nur eine moderate Änderung vorhandener Simulationscodes.
Direkter Vergleich bei vielen Arten von Anregungen
Um zu beurteilen, wie diese Methoden in der Praxis abschneiden, führte das Team umfangreiche Tests an einer breiten Palette molekularer Anregungen durch. Für niederenergetische angeregte Zustände in Benzol erreichte ΔCOOX konstant in weniger als etwa zehn Schritten eine Lösung, während ΔSCF-basierte Techniken manchmal viele Dutzend Schritte benötigten oder ganz scheiterten. Wenn ΔSCF jedoch zum beabsichtigten Zustand konvergierte, waren die vorhergesagten Anregungsenergien im Allgemeinen denen von ΔCOOX ähnlich. Systematische Vergleiche an Atomen und kleinen Molekülen zeigten, dass beide Ansätze experimentelle Werte gut für gewöhnliche Valenzanregungen, Rydberg-Zustände und echte Doppelanregungen, bei denen zwei Elektronen gefördert werden, erreichen können. ΔCOOX erwies sich jedoch als deutlich robuster: Es konvergierte selten zu einem falschen elektronischen Muster und blieb selbst bei herausfordernden Fällen mit schweren Atomen, starker Ladungsübertragung zwischen weit entfernten Fragmenten oder Molekülen in komplexer Umgebung stabil.
Komplexe Zustände und schwierige Umgebungen erreichen
Weil ΔCOOX die gewünschte Anregung durch seine Einschränkung erzwingt, garantiert es per Konstruktion, dass das elektronische Muster dem Ziel entspricht. Das macht es leichter zu erkennen, wenn eine Rechnung in die Irre gegangen ist, und erlaubt die Untersuchung gemischter Anregungen, die mehrere Orbitale gleichzeitig betreffen. Die Autoren zeigen außerdem, dass COOX sich natürlich erweitern lässt, um Moleküle in größeren Systemen zu behandeln, etwa ein kleines Gastmolekül in einer Fullerensäule oder ein Chromophor in einer proteinähnlichen Umgebung. In diesen Fällen wird der angeregte Zustand zuerst auf dem interessierenden Molekül definiert und dann nahtlos in die vollständige quantenmechanische Beschreibung der Umgebung eingebettet, wodurch die mühsame manuelle Orbitalverwaltung vermieden wird, die ΔSCF oft erfordert.
Was das für zukünftige Simulationen bedeutet
Die Studie kommt zu dem Schluss, dass die restriktionsbasierte ΔCOOX-Methode mindestens so genau ist wie etablierte ΔSCF-Schemata und in Bezug auf numerische Stabilität und Zuverlässigkeit oft deutlich überlegen ist. Für die meisten angeregten Zustände, die als ein einzelnes Elektron zwischen Orbitalen beschrieben werden können, erscheint ΔCOOX als starker Kandidat für das Standardwerkzeug und kann zudem als kluger Ausgangspunkt dienen, der älteren ΔSCF-Algorithmen beim Konvergieren hilft. Allgemeiner öffnet der vollständige COOX-Rahmen die Tür, routinemäßig komplizierte Anregungen und realistische chemische Umgebungen zu behandeln und rückt die Simulation angeregter Zustände näher an die unordentlichen, vielfältigen Systeme heran, die in realer Photochemie und Materialwissenschaft vorkommen.
Zitation: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Schlüsselwörter: angeregte Zustände, Dichtefunktionaltheorie, elektronische Struktur, Ladungsübertragung, rechnerische Chemie