Clear Sky Science · it
Un confronto dettagliato dei metodi ΔSCF con il metodo per stati eccitati a orbitali ottimizzati basato su vincoli
Perché questo è importante per la luce e le molecole
Quando le molecole assorbono luce, i loro elettroni vengono spinti in stati di maggiore energia, gli stati «eccitati». Predire con precisione queste eccitazioni è essenziale per comprendere celle solari, LED, fotocatalizzatori e persino il modo in cui la radiazione influenza i tessuti biologici. Questo articolo esamina in profondità due famiglie di metodi computazionali usati dai chimici per simulare elettroni eccitati, mostrando come un approccio più recente possa rendere questi calcoli sia più affidabili sia più versatili.
Come simaliamo normalmente gli elettroni eccitati
La maggior parte delle simulazioni moderne delle interazioni luce-materia usa un quadro di riferimento chiamato teoria del funzionale della densità dipendente dal tempo (TDDFT), che descrive come gli elettroni rispondono a piccole perturbazioni. È popolare perché è ragionevolmente accurata e relativamente economica in termini di risorse di calcolo. Però questo approccio standard fatica in alcuni dei casi più importanti: quando la carica si sposta su lunghe distanze, quando vengono eccitati elettroni interni (core) o quando sono eccitati più di un elettrone contemporaneamente. Per aggirare questi punti ciechi, i chimici si sono sempre più rivolti ai cosiddetti metodi a orbitali ottimizzati. Qui, uno stato eccitato viene costruito direttamente e quindi raffinato mediante una procedura auto-consistente, anziché essere dedotto come risposta dello stato fondamentale.
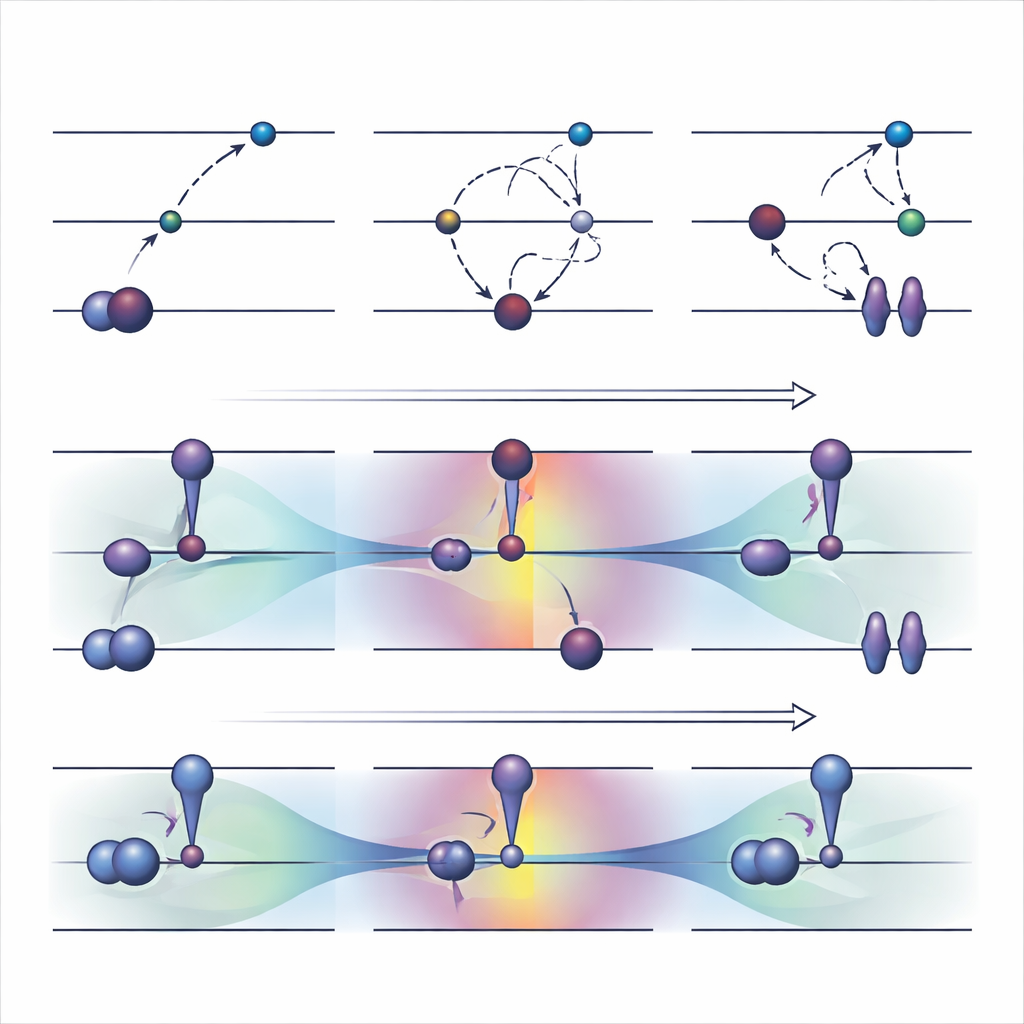
Strumenti tradizionali: forzare gli elettroni in nuovi posti
Una strategia a orbitali ottimizzati ampiamente usata, chiamata ΔSCF, funziona violando esplicitamente la regola usuale di riempimento degli elettroni: svuota un orbitale occupato e ne riempie uno di energia superiore, quindi riesegue l'ottimizzazione del sistema intorno a questo nuovo schema. Sulla base del ΔSCF di base, algoritmi pratici come il metodo della massima sovrapposizione cercano di impedire che il calcolo ricada nello stato fondamentale. Questi approcci possono descrivere eccitazioni difficili e hanno un costo computazionale approssimativamente pari a una normale esecuzione per lo stato fondamentale. Tuttavia, presentano svantaggi significativi. Spesso convergono lentamente o per nulla, possono silenziosamente ricadere nello stato sbagliato e sono in larga misura limitati a eccitazioni che appaiono come un semplice salto di un singolo elettrone tra due orbitali chiaramente identificabili.
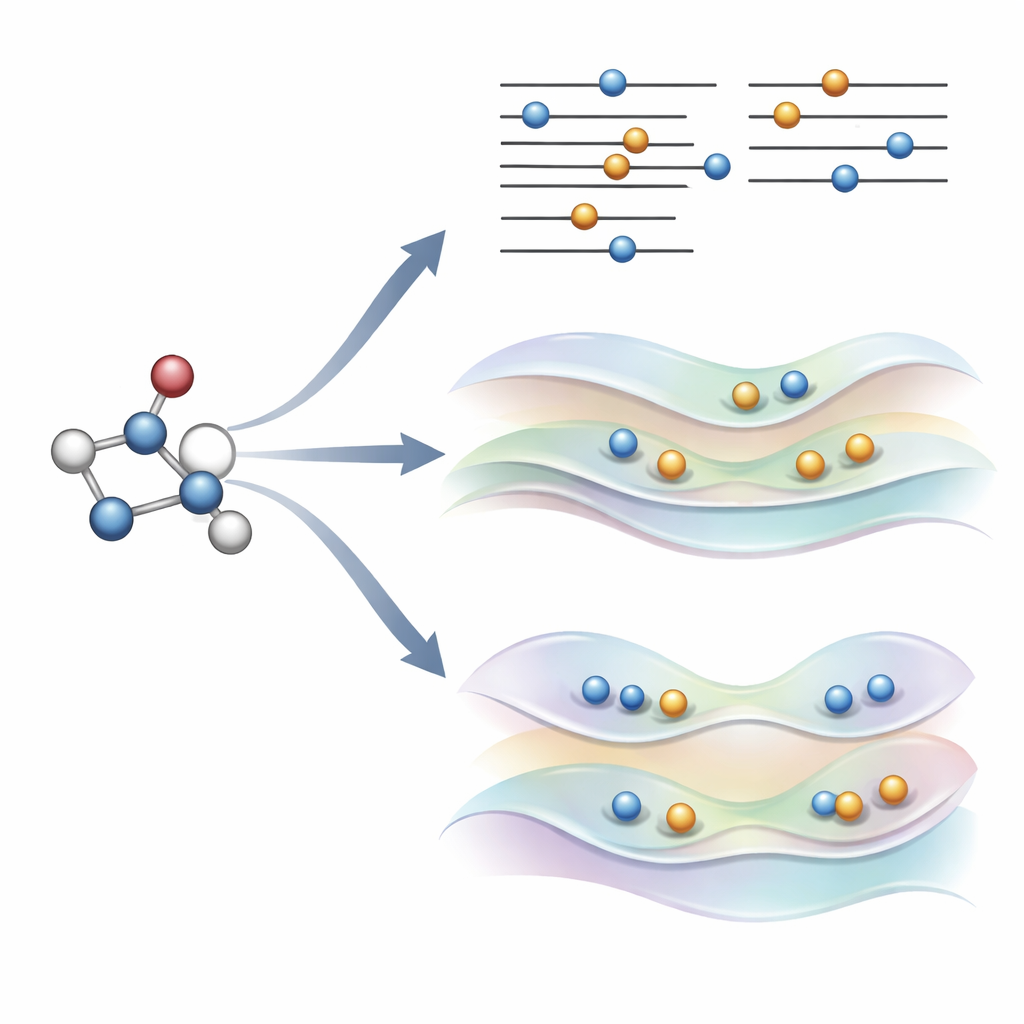
Una nuova idea: guidare delicatamente gli orbitali invece di infrangere le regole
Gli autori hanno recentemente proposto un'alternativa, chiamata COOX, che mantiene intatte le regole usuali di riempimento elettronico e aggiunge invece un vincolo appositamente studiato per spingere gli orbitali stessi verso una forma tipica dello stato eccitato. In questo studio si concentrano su una versione denominata ΔCOOX, concepita per imitare gli stessi semplici salti orbitali usati in ΔSCF così da poter confrontare direttamente i due approcci. Anziché cambiare manualmente le occupazioni, ΔCOOX aggiunge un potenziale supplementare che innalza o abbassa selettivamente l'energia di orbitali specifici fino a spostare effettivamente l'elettrone desiderato. Questo avviene all'interno del quadro flessibile della teoria del funzionale della densità vincolata e richiede solo una modifica modesta ai codici di simulazione esistenti.
Test affiancati su molti tipi di eccitazioni
Per giudicare le prestazioni pratiche di questi metodi, il team ha eseguito test estesi su un'ampia gamma di eccitazioni molecolari. Per stati eccitati a bassa energia nel benzene, ΔCOOX ha raggiunto costantemente una soluzione in meno di circa dieci passi, mentre le tecniche basate su ΔSCF a volte necessitavano di molte decine di passi o fallivano del tutto. Tuttavia, quando ΔSCF convergeva allo stato voluto, le energie di eccitazione previste erano generalmente simili a quelle di ΔCOOX. Confronti sistematici su atomi e piccole molecole hanno mostrato che entrambi gli approcci possono eguagliare bene i valori sperimentali per normali eccitazioni di valenza, stati di Rydberg e vere doppie eccitazioni dove sono promossi due elettroni. ΔCOOX si è però dimostrato molto più robusto: raramente è convergente verso lo schema elettronico sbagliato e rimane stabile anche per casi difficili che coinvolgono atomi pesanti, forti trasferimenti di carica tra frammenti distanti o molecole in ambienti complessi.
Raggiungere stati complicati e contesti complessi
Poiché ΔCOOX impone l'eccitazione desiderata tramite il suo vincolo, garantisce per costruzione che lo schema elettronico corrisponda all'obiettivo. Questo rende più facile riconoscere quando un calcolo è andato fuori strada ed esplorare eccitazioni miste che coinvolgono più orbitali contemporaneamente. Gli autori mostrano inoltre che COOX può essere naturalmente esteso per trattare molecole immerse in sistemi più grandi, come un piccolo ospite all'interno di una gabbia di fullerene o un cromoforo in un ambiente simile a una proteina. In questi casi, lo stato eccitato viene prima definito sulla molecola di interesse e poi integrato senza soluzione di continuità nella descrizione quantistica completa dell'ambiente circostante, evitando l'accurato ma laborioso conteggio manuale degli orbitali che ΔSCF spesso richiede.
Cosa significa per le simulazioni future
Lo studio conclude che il metodo ΔCOOX basato su vincoli è almeno altrettanto accurato quanto gli schemi ΔSCF consolidati ed è spesso chiaramente superiore in termini di stabilità numerica e affidabilità. Per la maggior parte degli stati eccitati descrivibili come il movimento di un singolo elettrone tra orbitali, ΔCOOX emerge come un forte candidato per diventare lo strumento predefinito, e può anche servire come punto di partenza intelligente che aiuta gli algoritmi ΔSCF più vecchi a convergere. Più in generale, il quadro completo COOX apre la strada a trattare routinariamente eccitazioni complesse e ambienti chimici realistici, avvicinando le simulazioni di stati eccitati ai sistemi disordinati e diversificati che si trovano nella fotochimica e nella scienza dei materiali del mondo reale.
Citazione: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Parole chiave: stati eccitati, teoria del funzionale della densità, struttura elettronica, trasferimento di carica, chimica computazionale