Clear Sky Science · ru
Детальное сравнение методов ΔSCF с методом оптимизации орбиталей для возбужденных состояний на основе ограничений
Почему это важно для света и молекул
Когда молекулы поглощают свет, их электроны переходят в состояния с более высокой энергией — «возбужденные» состояния. Точное предсказание этих возбуждений необходимо для понимания работы солнечных батарей, светодиодов, фотокатализаторов и даже влияния излучения на биологические ткани. В этой статье исследуются две группы компьютерных методов, которыми химики моделируют возбужденные электроны, и показано, как более новый подход может сделать такие расчеты одновременно более надежными и более универсальными.
Как мы обычно моделируем возбужденные электроны
Большинство современных симуляций взаимодействия света с веществом используют рабочую лошадку — теорию функционала плотности во временной области (TD-DFT), которая описывает, как электроны реагируют на малые возмущения. Она популярна, потому что дает разумную точность при относительно невысокой вычислительной стоимости. Но этот стандартный подход испытывает затруднения в некоторых ключевых ситуациях: при переносе заряда на большие расстояния, при возбуждении внутренних (ядерных) электронов или когда возбуждается более одного электрона одновременно. Чтобы обойти эти «слепые зоны», химики всё чаще обращаются к так называемым методам с оптимизацией орбиталей. В них возбужденное состояние строится напрямую и затем уточняется самосогласованной процедурой, а не выводится как возмущение основного состояния.
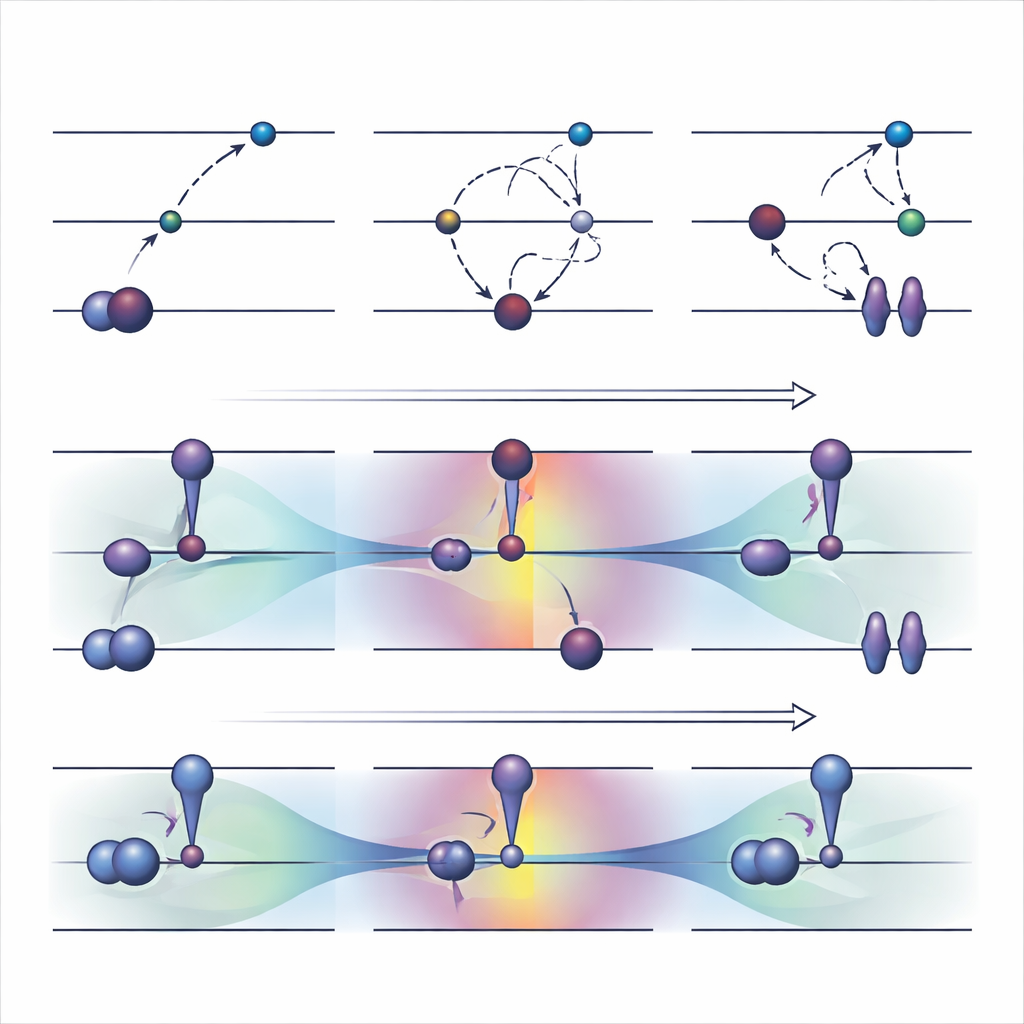
Старые инструменты: принудительное переселение электронов
Широко используемая стратегия с оптимизацией орбиталей, называемая ΔSCF, работает путем явного нарушения обычного правила заполнения электронов: освобождается занятая орбиталь и заполняется более высокая, после чего система реоптимизируется вокруг этой новой конфигурации. Поверх базового ΔSCF практические алгоритмы, такие как метод максимального перекрытия, пытаются не дать расчету «соскользнуть» обратно в основное состояние. Эти подходы способны описывать трудные возбуждения и по стоимости примерно сравнимы с обычным расчётом основного состояния. Однако у них есть существенные недостатки. Они часто сходятся медленно или вовсе не сходятся, могут незаметно возвращаться к неверному состоянию и в значительной степени ограничены возбуждениями, которые выглядят как простой переход одного электрона между двумя явно определёнными орбиталями.
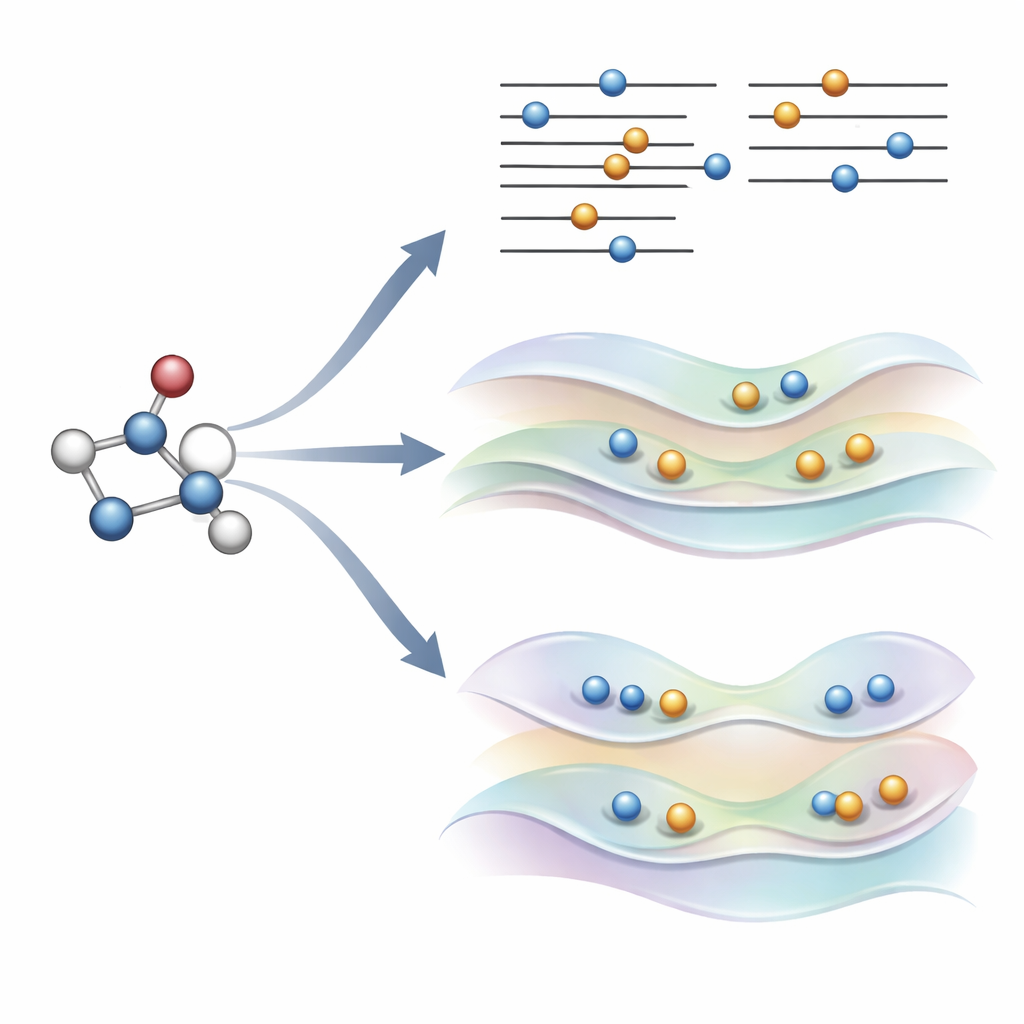
Новая идея: осторожно направлять орбитали вместо нарушения правил
Авторы недавно предложили альтернативу под названием COOX, которая сохраняет обычные правила заполнения электронов и вместо этого вводит специально сконструированное ограничение, чтобы «подтолкнуть» сами орбитали к форме возбужденного состояния. В этом исследовании они сосредоточились на варианте, обозначенном ΔCOOX, который спроектирован так, чтобы имитировать те же простые орбитальные переходы, что и в ΔSCF, для прямого сравнения двух подходов. Вместо ручного изменения заполнений, ΔCOOX добавляет дополнительный потенциал, который выборочно повышает или понижает энергию конкретных орбиталей до тех пор, пока требуемый электрон фактически не переместится. Это выполняется в гибкой рамке ограниченной теории функционала плотности и требует лишь небольшой модификации существующих кодов симуляции.
Параллельные тесты на разных типах возбуждений
Чтобы оценить практическую эффективность методов, исследователи провели обширные тесты на широком наборе молекулярных возбуждений. Для низколежащих возбужденных состояний бензола ΔCOOX последовательно находил решение за менее чем примерно десять шагов, в то время как техники на основе ΔSCF иногда требовали десятков шагов или вовсе давали сбой. Тем не менее, когда ΔSCF сходился к требуемому состоянию, предсказанные энергии возбуждения в целом были схожи с результатами ΔCOOX. Систематические сравнения на атомах и малых молекулах показали, что оба подхода могут хорошо согласовываться с экспериментом для обычных валентных возбуждений, rydberg‑состояний и действительно двойных возбуждений, где продвигаются два электрона. Однако ΔCOOX оказался гораздо более робастным: он редко сходился к неверной электронной конфигурации и оставался стабильным даже в сложных случаях с тяжёлыми атомами, сильным переносом заряда между удалёнными фрагментами или молекулами в сложной среде.
Достижение хитрых состояний и сложной окружающей среды
Поскольку ΔCOOX принуждает к нужному возбуждению через своё ограничение, он по конструкции гарантирует, что электронная конфигурация соответствует цели. Это упрощает распознавание случаев, когда расчет ушёл в сторону, и позволяет исследовать смешанные возбуждения, включающие несколько орбиталей одновременно. Авторы также показывают, что COOX естественно расширяется на случаи, когда молекула находится в более крупной среде, например маленький гость внутри фуллереновой клетки или хромофор в «белкообразной» среде. В таких ситуациях возбужденное состояние сначала определяется для самой молекулы, а затем бесшовно встраивается в полное квантовое описание окружения, избегая утомительного ручного учёта орбиталей, который часто требуется в ΔSCF.
Что это означает для будущих симуляций
Авторы приходят к выводу, что метод ΔCOOX на основе ограничений по крайней мере не уступает проверенным схемам ΔSCF по точности и часто явно превосходит их по численной стабильности и надёжности. Для большинства возбужденных состояний, которые можно описать как переход одного электрона между орбиталями, ΔCOOX выступает сильным кандидатом на роль инструмента по умолчанию и может также служить разумной отправной точкой, помогающей старым алгоритмам ΔSCF сходиться. В более широком смысле полная рамка COOX открывает возможность рутинной обработки сложных возбуждений и реалистичных химических окружений, приближая моделирование возбуждённых состояний к запутанным, разнообразным системам, встречающимся в реальной фотохимии и материаловедении.
Цитирование: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Ключевые слова: возбужденные состояния, теория функционала плотности, электронная структура, перенос заряда, вычислительная химия