Clear Sky Science · fr
Une comparaison détaillée des méthodes ΔSCF avec la méthode de l’état excité optimisée orbitalement basée sur des contraintes
Pourquoi cela compte pour la lumière et les molécules
Lorsque les molécules absorbent de la lumière, leurs électrons sont poussés vers des états de plus haute énergie dits « excités ». Prédire ces excitations avec précision est essentiel pour comprendre les cellules solaires, les LED, les photocatalyseurs et même la façon dont les radiations affectent les tissus biologiques. Cet article examine en profondeur deux familles de méthodes informatiques que les chimistes utilisent pour simuler les électrons excités, et montre comment une approche plus récente peut rendre ces calculs à la fois plus fiables et plus polyvalents.
Comment nous simulons habituellement les électrons excités
La plupart des simulations modernes des interactions lumière-matière utilisent un cadre de référence appelé théorie de la fonctionnelle de la densité dépendante du temps (TDDFT), qui traite de la réponse des électrons à de petites perturbations. Elle est prisée parce qu’elle offre une précision raisonnable tout en restant relativement peu coûteuse en calcul. Mais cette approche standard peine sur certains des cas les plus importants : lorsque la charge est déplacée sur de longues distances, lorsque des électrons internes (de cœur) sont excités, ou lorsque plusieurs électrons sont excités simultanément. Pour contourner ces angles morts, les chimistes se tournent de plus en plus vers des méthodes dites optimisées orbitalement. Là, un état excité est construit directement puis affiné par une procédure d’auto-cohérence, au lieu d’être déduit comme la réponse de l’état fondamental.
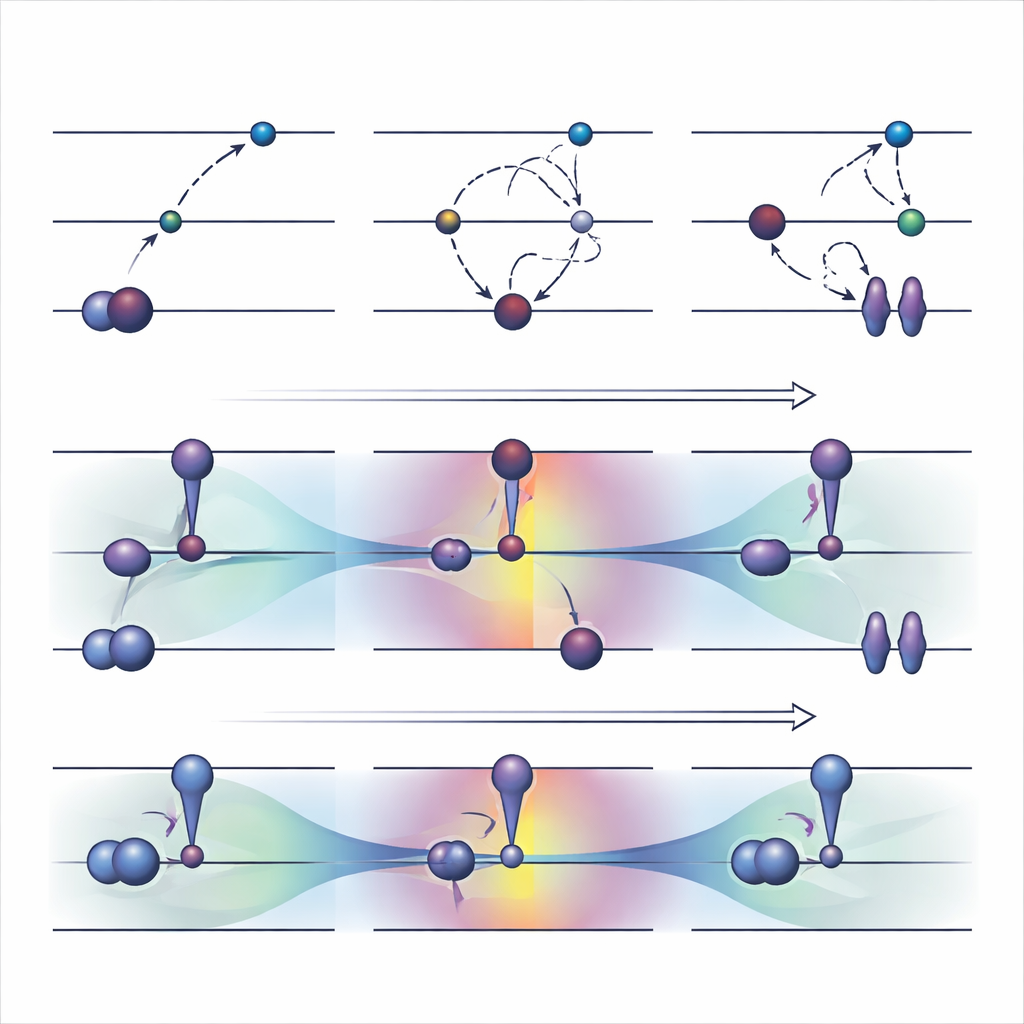
Outils anciens : forcer les électrons dans de nouvelles cases
Une stratégie optimisée orbitalement largement utilisée, appelée ΔSCF, fonctionne en violant explicitement la règle habituelle d’occupation des électrons : elle vide une orbitale occupée et remplit une orbitale de plus haute énergie, puis réoptimise le système autour de ce nouvel agencement. Par-dessus le ΔSCF de base, des algorithmes pratiques comme la méthode du recouvrement maximal tentent d’empêcher le calcul de retomber vers l’état fondamental. Ces approches peuvent décrire des excitations difficiles et coûtent à peu près autant qu’un calcul normal d’état fondamental. Cependant, elles présentent des inconvénients sérieux. Elles convergent souvent lentement voire pas du tout, peuvent silencieusement revenir à l’état incorrect, et sont largement limitées aux excitations qui ressemblent à un simple saut d’un seul électron entre deux orbitales clairement identifiables.
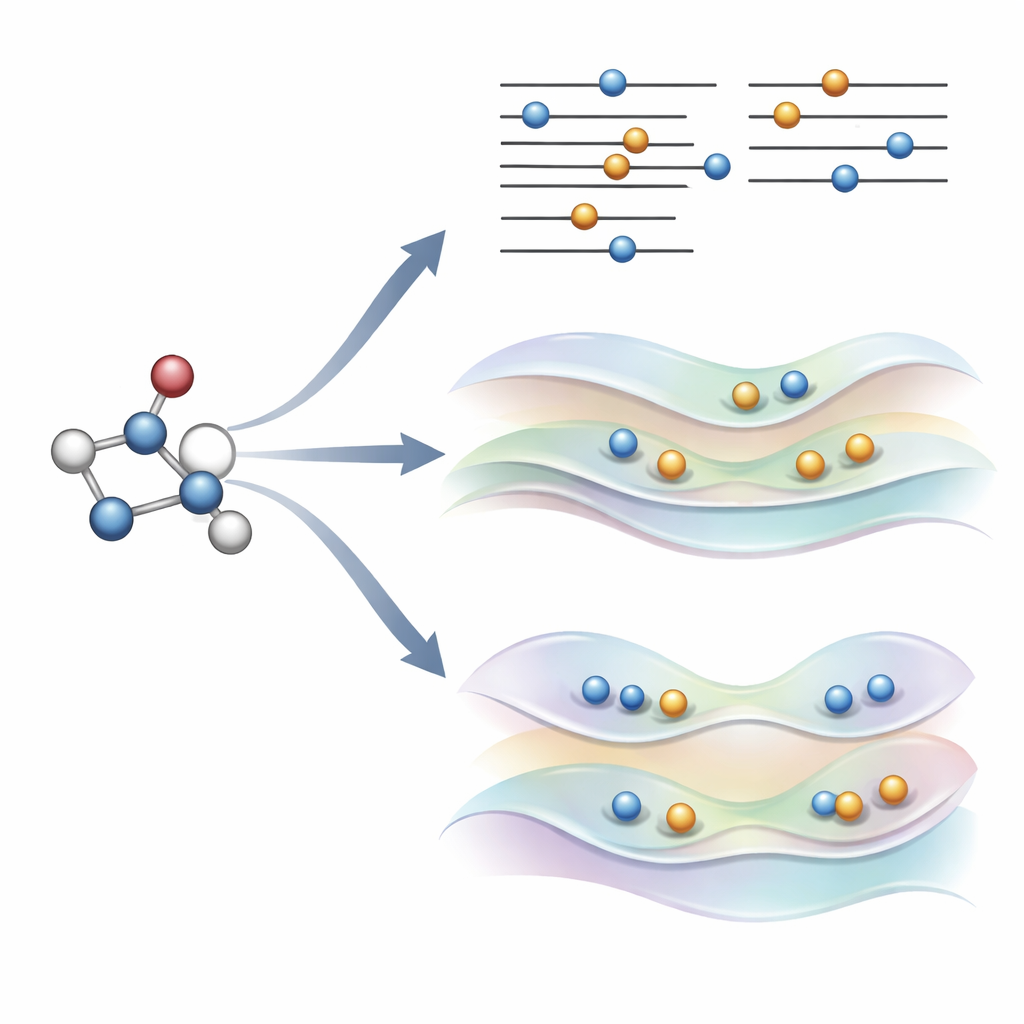
Une idée nouvelle : orienter doucement les orbitales au lieu de briser les règles
Les auteurs ont récemment proposé une alternative, appelée COOX, qui conserve les règles habituelles d’occupation électronique et ajoute à la place une contrainte soigneusement conçue pour pousser les orbitales elles-mêmes vers une forme d’état excité. Dans cette étude, ils se concentrent sur une version baptisée ΔCOOX, conçue pour imiter les mêmes sauts orbitaux simples utilisés en ΔSCF afin que les deux approches puissent être comparées directement. Plutôt que de changer manuellement les occupations, ΔCOOX ajoute un potentiel supplémentaire qui élève ou abaisse sélectivement l’énergie d’orbitales spécifiques jusqu’à ce que l’électron désiré soit effectivement déplacé. Ceci s’effectue dans le cadre flexible de la théorie de la fonctionnelle de la densité contrainte, et ne nécessite qu’une modification modeste des codes de simulation existants.
Tests côte à côte sur de nombreux types d’excitations
Pour juger de la performance de ces méthodes en pratique, l’équipe a réalisé des tests étendus sur une large gamme d’excitations moléculaires. Pour les états excités de faible énergie dans le benzène, ΔCOOX a systématiquement atteint une solution en moins d’une dizaine d’itérations, tandis que les techniques basées sur ΔSCF nécessitaient parfois plusieurs dizaines d’itérations ou échouaient complètement. Pourtant, lorsque ΔSCF convergea vers l’état visé, les énergies d’excitation prédites étaient généralement proches de celles de ΔCOOX. Des comparaisons systématiques sur des atomes et de petites molécules ont montré que les deux approches peuvent bien reproduire les valeurs expérimentales pour les excitations de valence ordinaires, les états Rydberg et les véritables doubles excitations où deux électrons sont promus. Cependant, ΔCOOX s’est révélé beaucoup plus robuste : il convergeait rarement vers un mauvais agencement électronique et restait stable même pour des cas difficiles impliquant des atomes lourds, un transfert de charge intense entre des fragments éloignés, ou des molécules dans des environnements complexes.
Atteindre des états délicats et des environnements complexes
Parce que ΔCOOX impose l’excitation souhaitée via sa contrainte, il garantit par construction que le schéma électronique correspond bien à la cible. Cela facilite la détection des calculs qui ont dévié et permet d’explorer des excitations mixtes impliquant plusieurs orbitales à la fois. Les auteurs montrent également que COOX peut être étendu naturellement pour traiter des molécules intégrées dans des systèmes plus larges, comme un petit hôte à l’intérieur d’une cage de fullerène ou un chromophore dans un environnement de type protéine. Dans ces cas, l’état excité est d’abord défini sur la molécule d’intérêt puis intégré de manière fluide dans la description quantique complète de l’environnement, évitant la tenue manuelle et laborieuse des occupations orbitales que ΔSCF exige souvent.
Ce que cela signifie pour les simulations futures
L’étude conclut que la méthode ΔCOOX basée sur des contraintes est au moins aussi précise que les schémas ΔSCF établis et dépasse souvent ces derniers en termes de stabilité numérique et de fiabilité. Pour la plupart des états excités qui peuvent être décrits comme le déplacement d’un seul électron entre orbitales, ΔCOOX émerge comme un candidat solide pour devenir l’outil par défaut, et il peut aussi servir de point de départ intelligent aidant les anciens algorithmes ΔSCF à converger. Plus largement, le cadre complet COOX ouvre la porte au traitement de routine d’excitations compliquées et d’environnements chimiques réalistes, rapprochant les simulations d’états excités des systèmes désordonnés et variés que l’on trouve en photochimie et en science des matériaux dans le monde réel.
Citation: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Mots-clés: états excités, théorie de la fonctionnelle de la densité, structure électronique, transfert de charge, chimie computationnelle