Clear Sky Science · en
A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method
Why this matters for light and molecules
When molecules absorb light, their electrons are pushed into higher-energy "excited" states. Accurately predicting these excitations is essential for understanding solar cells, LEDs, photocatalysts, and even the way radiation affects biological tissue. This paper looks under the hood of two families of computer methods that chemists use to simulate excited electrons, and shows how a newer approach can make these calculations both more reliable and more versatile.
How we usually simulate excited electrons
Most modern simulations of light–matter interactions use a workhorse framework called time-dependent density functional theory, which treats how electrons respond to small disturbances. It is popular because it is reasonably accurate and relatively cheap to run on a computer. But this standard approach struggles with some of the most important cases: when charge is moved over long distances, when inner (core) electrons are excited, or when more than one electron is excited at once. To get around these blind spots, chemists have increasingly turned to so-called orbital-optimized methods. Here, an excited state is built directly and then refined by a self-consistent procedure, rather than inferred as a response of the ground state.
Old tools: forcing electrons into new slots
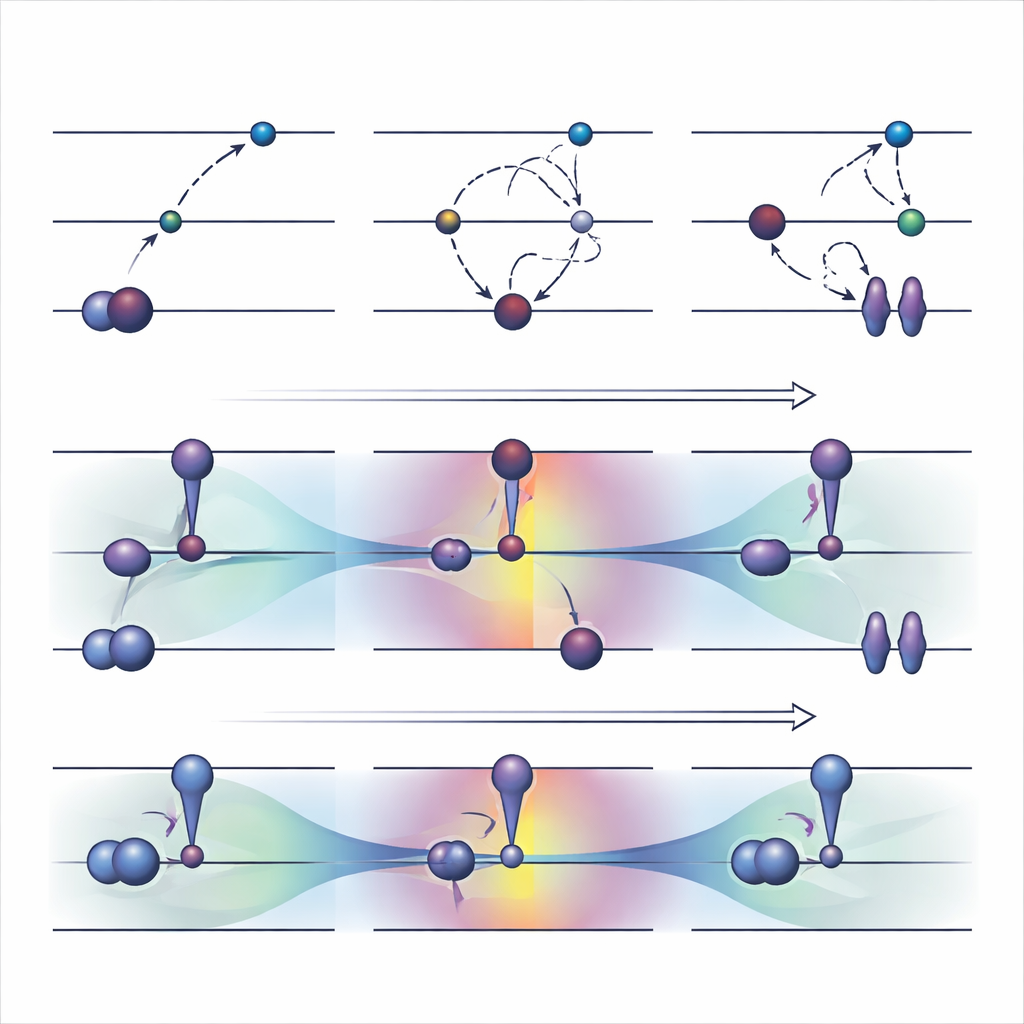
A widely used orbital-optimized strategy, called ΔSCF, works by explicitly violating the usual filling rule for electrons: it empties an occupied orbital and fills a higher one, then re-optimizes the system around this new pattern. On top of basic ΔSCF, practical algorithms like the maximum overlap method try to keep the calculation from slipping back to the ground state. These approaches can describe difficult excitations and cost roughly the same as a normal ground-state run. However, they come with serious drawbacks. They often converge slowly or not at all, can quietly fall back to the wrong state, and are largely limited to excitations that look like a simple single-electron jump between two clearly identified orbitals.
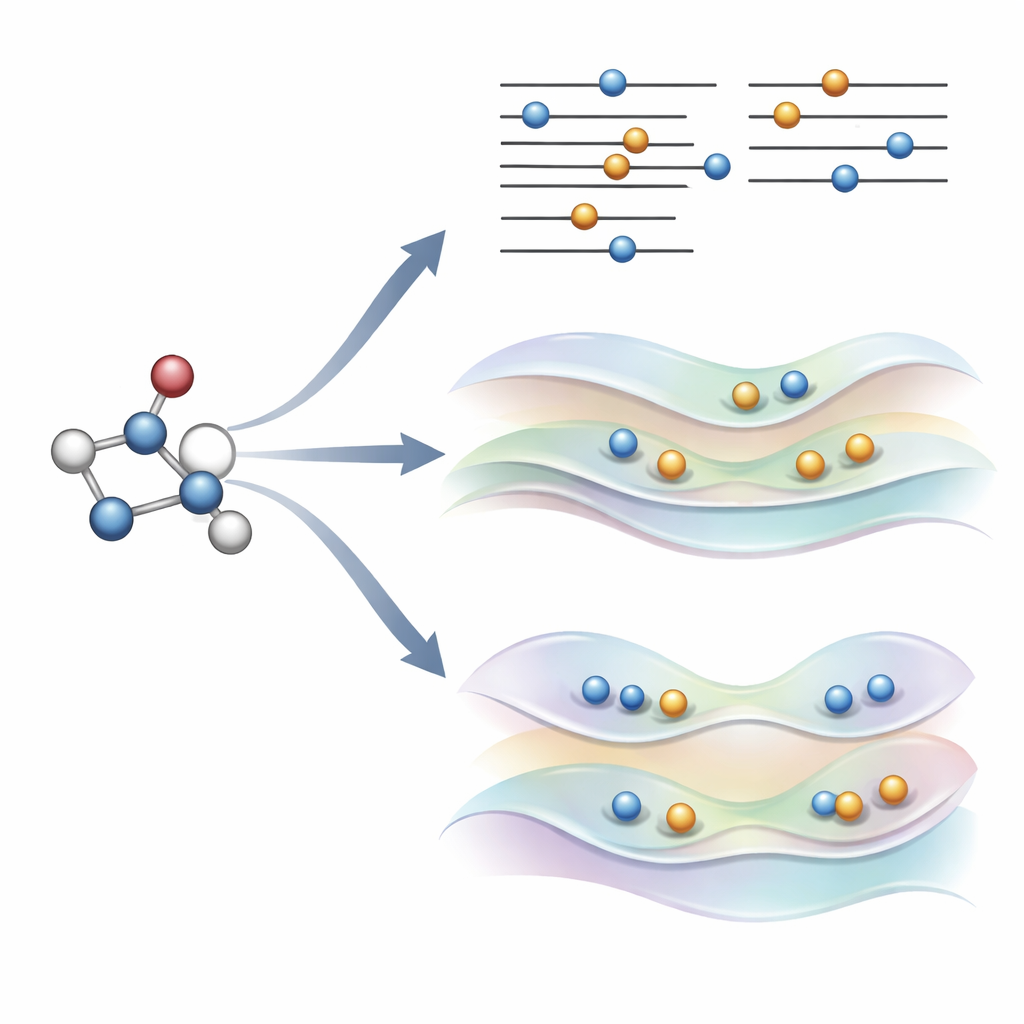
A new idea: gently steering orbitals instead of breaking rules
The authors recently proposed an alternative, called COOX, that keeps the usual electron-filling rules intact and instead adds a carefully designed constraint to push the orbitals themselves into an excited-state shape. In this study they focus on a version dubbed ΔCOOX, which is built to mimic the same simple orbital jumps used in ΔSCF so that the two approaches can be compared directly. Rather than manually changing occupations, ΔCOOX adds an extra potential that selectively raises and lowers the energy of specific orbitals until the desired electron is effectively moved. This is done within the flexible framework of constrained density functional theory, and requires only a modest modification to existing simulation codes.
Side-by-side tests on many kinds of excitations
To judge how these methods perform in practice, the team ran extensive tests on a wide range of molecular excitations. For low-lying excited states in benzene, ΔCOOX consistently reached a solution in fewer than about ten steps, while ΔSCF-based techniques sometimes needed many tens of steps or failed altogether. Yet when ΔSCF did converge to the intended state, the predicted excitation energies were generally similar to those from ΔCOOX. Systematic comparisons on atoms and small molecules showed that both approaches can match experimental values well for ordinary valence excitations, Rydberg states, and true double excitations where two electrons are promoted. However, ΔCOOX proved much more robust: it rarely converged to the wrong electronic pattern and remained stable even for challenging cases involving heavy atoms, strong charge transfer between distant fragments, or molecules in complex environments.
Reaching tricky states and complex surroundings
Because ΔCOOX enforces the desired excitation through its constraint, it guarantees that the electronic pattern matches the target by construction. This makes it easier to recognize when a calculation has gone astray and to explore mixed excitations that involve several orbitals at once. The authors also show that COOX can be naturally extended to treat molecules embedded in larger systems, such as a small guest inside a fullerene cage or a chromophore inside a protein-like environment. In these cases, the excited state is first defined on the molecule of interest and then seamlessly embedded into the full quantum description of the surroundings, avoiding the tedious manual orbital bookkeeping that ΔSCF often requires.
What this means for future simulations
The study concludes that the constraint-based ΔCOOX method is at least as accurate as established ΔSCF schemes and is often clearly superior in terms of numerical stability and reliability. For most excited states that can be described as a single electron moving between orbitals, ΔCOOX emerges as a strong candidate for the default tool of choice, and it can also serve as a smart starting point that helps older ΔSCF algorithms converge. More broadly, the full COOX framework opens the door to routinely treating complicated excitations and realistic chemical environments, pushing excited-state simulations closer to the messy, diverse systems found in real-world photochemistry and materials science.
Citation: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Keywords: excited states, density functional theory, electronic structure, charge transfer, computational chemistry