Clear Sky Science · ja
制約付き軌道最適化励起状態法とΔSCF法の詳細な比較
光と分子にとってこれはなぜ重要か
分子が光を吸収すると、電子はより高いエネルギーの「励起」状態へ押し上げられます。これらの励起を正確に予測することは、太陽電池、LED、光触媒、さらには放射線が生体組織に与える影響を理解するうえで不可欠です。本論文は、化学者が励起電子をシミュレートするのに用いる二つの計算手法群の内部を詳しく検証し、新しいアプローチがどのように計算をより信頼性が高く多用途にできるかを示します。
通常、励起電子をどうシミュレートするか
光–物質相互作用のほとんどの現代的シミュレーションは、電子が小さな摂動にどう応答するかを扱う定番フレームワークである時間依存密度汎関数理論(TDDFT)を使用します。これは比較的精度が高く計算コストも抑えられるため広く用いられています。しかし、この標準的手法は重要なケース、たとえば電荷が長距離移動する場合、内殻(コア)電子が励起される場合、あるいは複数の電子が同時に励起される場合などで苦手とします。これらの盲点を回避するために、化学者たちは軌道最適化法と呼ばれる手法にますます頼るようになっています。ここでは励起状態を応答として基底状態から推定するのではなく、直接構築して自己無撞着な手続きで精緻化します。
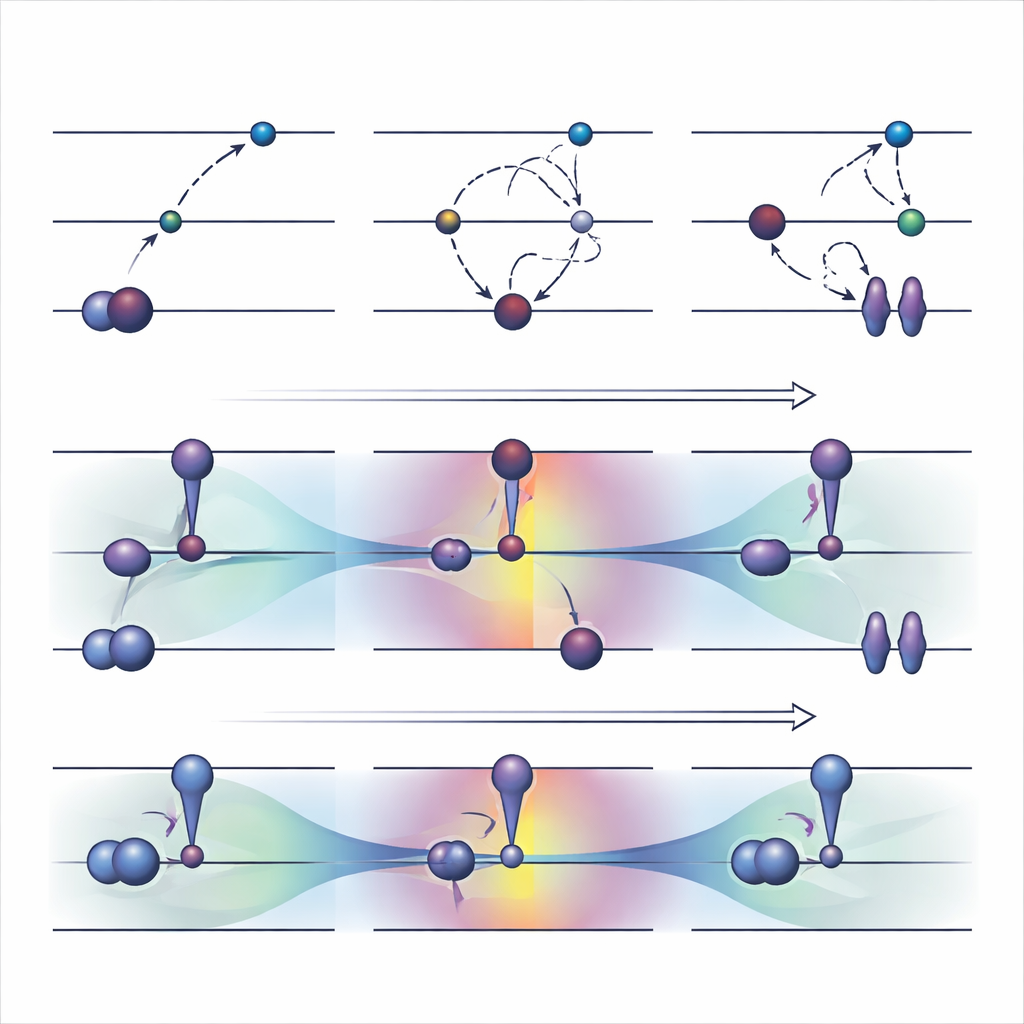
古い道具:電子を無理やり別のスロットへ
広く使われている軌道最適化戦略の一つであるΔSCFは、電子の通常の占有規則を明示的に破ることで動作します:占有された軌道を空にし、より高い軌道を占有させ、その新しい占有パターンの周りで系を再最適化します。基本的なΔSCFの上で、最大重なり法などの実用的アルゴリズムは計算が基底状態へ戻ってしまうのを防ごうとします。これらの手法は扱いにくい励起を記述でき、計算コストは通常の基底状態計算とほぼ同程度です。しかし深刻な欠点もあります。収束が非常に遅いか、まったく収束しないことがあり、目的とは異なる状態へひそかに落ち込むことがあり、また明確に同定できる二つの軌道間の単純な一電子遷移のような励起に大きく制限されます。
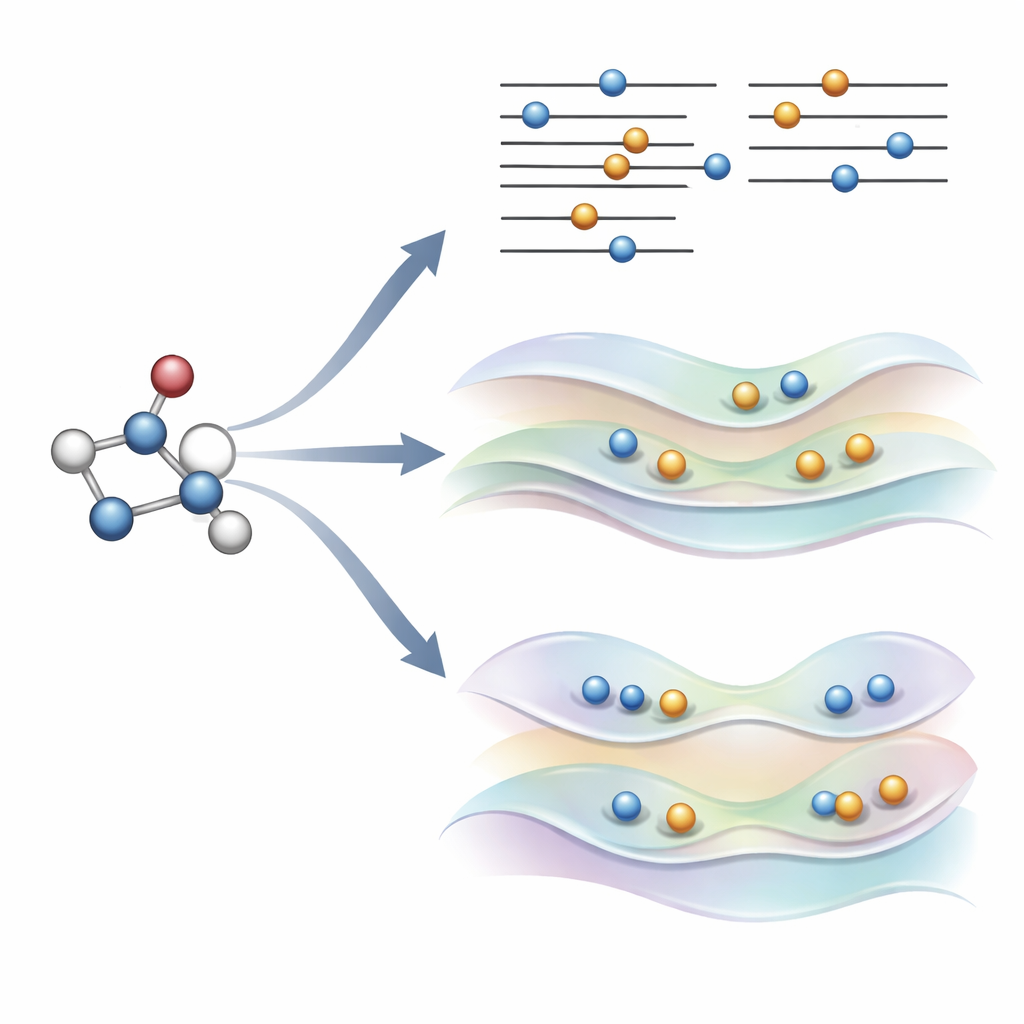
新しい考え方:規則を破る代わりに軌道をやんわり導く
著者らは最近、COOXと呼ばれる代替手法を提案しました。これは従来の電子占有規則を維持しつつ、精巧に設計された制約を加えて軌道自体を励起状態の形状へと押しやるものです。本研究ではΔSCFと直接比較できるよう同じ単純な軌道遷移を模倣するように構築されたΔCOOXというバージョンに焦点を当てています。占有を手動で変更する代わりに、ΔCOOXは特定の軌道のエネルギーを選択的に上げ下げする余分なポテンシャルを加え、目的とする電子が実質的に移動するまで調整します。これは制約付き密度汎関数理論の柔軟な枠組み内で行われ、既存のシミュレーションコードへの修正は比較的軽微です。
さまざまな励起に対する並列テスト
これらの手法の実用的な性能を評価するために、研究チームは幅広い分子励起について大規模なテストを実行しました。ベンゼンの低エネルギー励起状態では、ΔCOOXは一貫して約十ステップ未満で解に到達したのに対し、ΔSCFベースの技法は数十ステップを要するか完全に失敗することがありました。しかしΔSCFが目的の状態に収束した場合、予測される励起エネルギーは概してΔCOOXのものと類似していました。原子や小分子に対する体系的比較では、通常の価電子励起、リュードベリ状態、二電子が同時に励起される真の二重励起について両アプローチが実験値とよく一致することが示されました。とはいえ、ΔCOOXははるかに堅牢であることが判明しました:誤った電子配置に収束することがほとんどなく、重い原子を含む場合、遠く離れた断片間の強い電荷移動、あるいは複雑な環境に置かれた分子といった困難なケースでも安定していました。
到達が難しい状態や複雑な環境への対応
ΔCOOXは制約によって目的の励起を強制するため、電子配置が設計どおりであることを構成的に保証します。これにより、計算が逸脱したことを認識しやすくなり、複数の軌道が関与する混合励起を探ることも容易になります。著者らはまた、COOXがフラーレンケージ内の小さなゲストやタンパク質様環境内のクロモフォアなど、より大きな系に埋め込まれた分子を扱うよう自然に拡張できることを示しています。これらの場合、励起状態はまず対象分子上で定義され、その後周囲の量子記述へとシームレスに埋め込まれるため、ΔSCFがしばしば必要とする面倒な手作業による軌道の管理を回避できます。
今後のシミュレーションにとっての意味
研究は、制約ベースのΔCOOX法が従来のΔSCF方式と同等以上の精度を持ち、数値的安定性と信頼性の面でしばしば明確に優れていると結論づけています。軌道間を移動する単一電子で記述できるほとんどの励起状態に対して、ΔCOOXはデフォルトの有力な候補として浮上し、また古いΔSCFアルゴリズムの収束を助ける賢い出発点にもなりえます。より広い視点では、COOXのフルフレームワークは複雑な励起や現実的な化学環境を日常的に扱う道を開き、励起状態シミュレーションを実世界の光化学や材料科学で見られる混沌で多様な系に近づけます。
引用: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
キーワード: 励起状態, 密度汎関数理論, 電子構造, 電荷移動, 計算化学