Clear Sky Science · pt
Uma comparação detalhada dos métodos ΔSCF com o método de estados excitados otimizado por orbitais baseado em restrições
Por que isso importa para luz e moléculas
Quando moléculas absorvem luz, seus elétrons são impulsionados para estados de maior energia, chamados "excitados". Prever essas excitações com precisão é essencial para entender células solares, LEDs, fotocatalisadores e até a forma como a radiação afeta tecido biológico. Este artigo examina em detalhe duas famílias de métodos computacionais que químicos usam para simular elétrons excitados e mostra como uma abordagem mais recente pode tornar esses cálculos tanto mais confiáveis quanto mais versáteis.
Como normalmente simulamos elétrons excitados
A maioria das simulações modernas de interação luz–matéria usa uma estrutura de trabalho chamada teoria do funcional da densidade dependente do tempo (TDDFT), que trata de como os elétrons respondem a perturbações pequenas. É popular porque oferece precisão razoável a um custo computacional relativamente baixo. Mas essa abordagem padrão enfrenta dificuldades em alguns dos casos mais importantes: quando a carga é deslocada por longas distâncias, quando elétrons internos (de núcleo) são excitados ou quando mais de um elétron é excitado simultaneamente. Para contornar esses pontos cegos, químicos têm recorrido cada vez mais a métodos chamados de otimização orbital. Nessa classe, um estado excitado é construído diretamente e então refinado por um procedimento autoconsistente, em vez de ser inferido como uma resposta do estado fundamental.
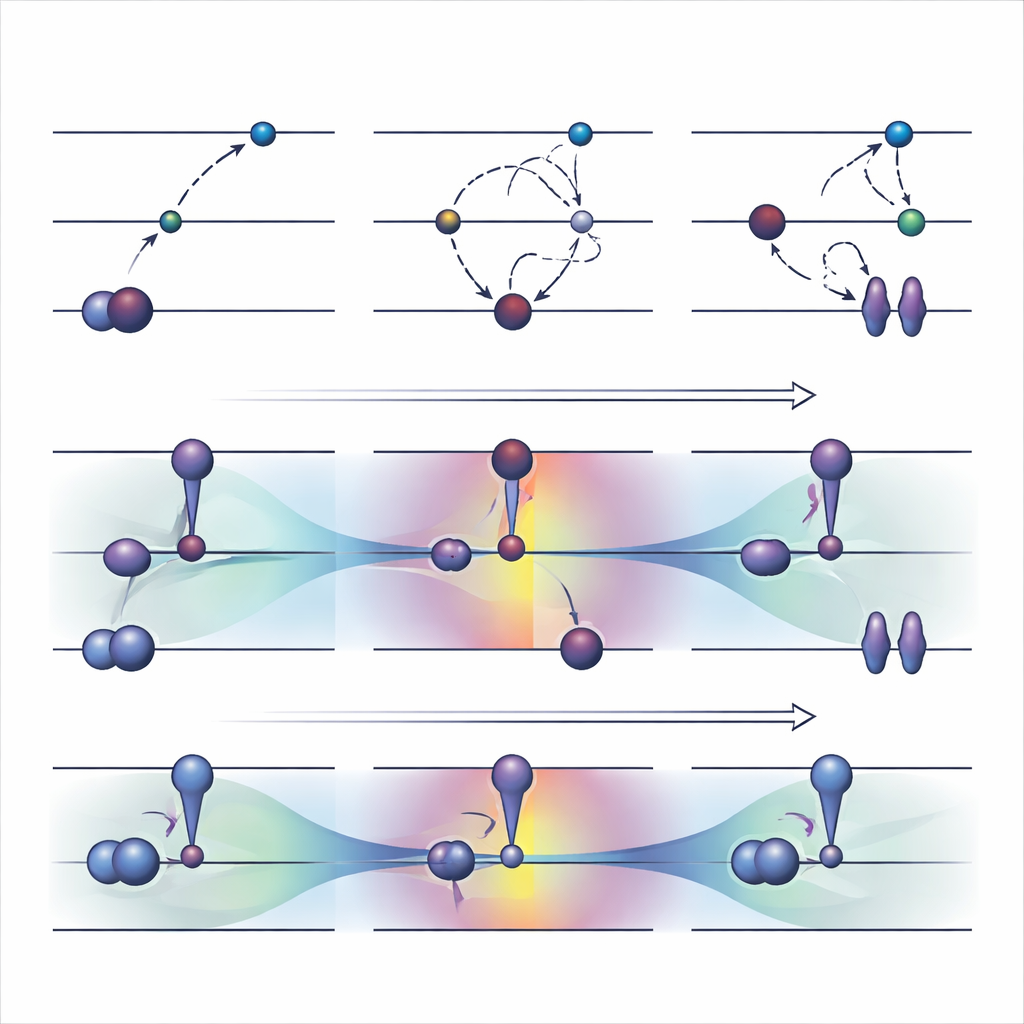
Ferramentas antigas: forçando elétrons a novos níveis
Uma estratégia amplamente usada de otimização orbital, chamada ΔSCF, funciona violando explicitamente a regra usual de preenchimento de elétrons: esvazia-se um orbital ocupado e preenche-se um orbital de maior energia, depois re-optimiza-se o sistema em torno desse novo padrão. Além do ΔSCF básico, algoritmos práticos como o método de máxima sobreposição tentam impedir que o cálculo retorne ao estado fundamental. Essas abordagens conseguem descrever excitações difíceis e custam, em geral, aproximadamente o mesmo que uma execução normal do estado fundamental. Contudo, vêm com desvantagens sérias. Frequentemente convergem de forma lenta ou não convergem, podem silenciosamente regressar ao estado errado e são em grande parte limitadas a excitações que se parecem com um simples salto de um único elétron entre dois orbitais claramente identificáveis.
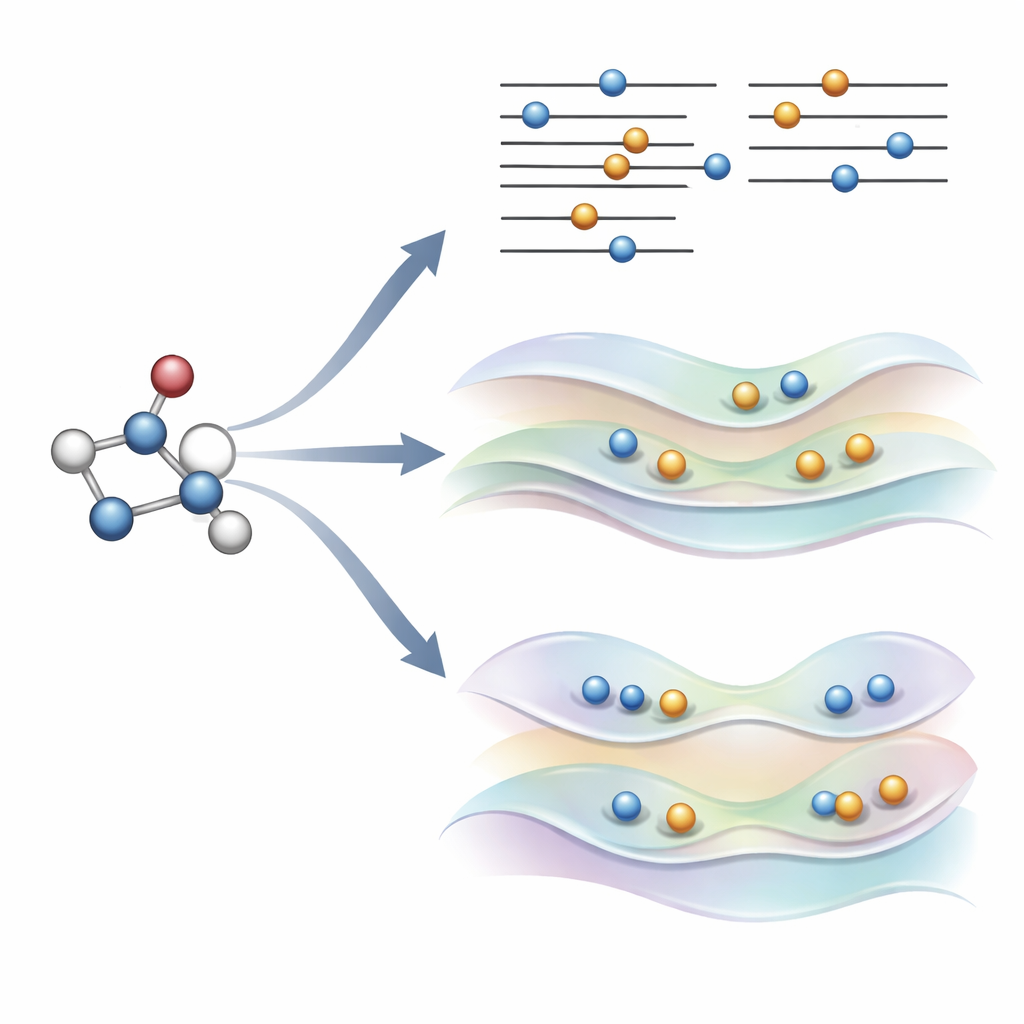
Uma ideia nova: orientar suavemente os orbitais em vez de quebrar regras
Os autores propuseram recentemente uma alternativa, chamada COOX, que mantém as regras usuais de preenchimento eletrônico e adiciona uma restrição cuidadosamente desenhada para empurrar os próprios orbitais a uma configuração de estado excitado. Neste estudo eles se concentram em uma versão apelidada ΔCOOX, projetada para imitar os mesmos saltos orbitais simples usados no ΔSCF, de modo que as duas abordagens possam ser comparadas diretamente. Em vez de mudar manualmente as ocupações, o ΔCOOX adiciona um potencial extra que eleva e abaixa seletivamente a energia de orbitais específicos até que o elétron desejado seja efetivamente deslocado. Isso é feito dentro da estrutura flexível da teoria do funcional da densidade com restrições e requer apenas uma modesta modificação nos códigos de simulação existentes.
Testes lado a lado em muitos tipos de excitações
Para avaliar como esses métodos se comportam na prática, a equipe realizou testes extensivos em uma grande variedade de excitações moleculares. Para estados excitados de baixa energia em benzeno, o ΔCOOX atingiu consistentemente uma solução em menos de cerca de dez passos, enquanto técnicas baseadas em ΔSCF às vezes precisaram de muitas dezenas de passos ou falharam completamente. Ainda assim, quando o ΔSCF convergia para o estado pretendido, as energias de excitação previstas eram geralmente similares às do ΔCOOX. Comparações sistemáticas em átomos e pequenas moléculas mostraram que ambas as abordagens podem coincidir bem com valores experimentais para excitações de valência comuns, estados de Rydberg e verdadeiras dupla excitações em que dois elétrons são promovidos. No entanto, o ΔCOOX mostrou-se muito mais robusto: raramente convergiu para o padrão eletrônico errado e manteve-se estável mesmo em casos desafiadores envolvendo átomos pesados, forte transferência de carga entre fragmentos distantes ou moléculas em ambientes complexos.
Atingindo estados difíceis e ambientes complexos
Porque o ΔCOOX impõe a excitação desejada por meio de sua restrição, garante-se por construção que o padrão eletrônico corresponde ao alvo. Isso facilita reconhecer quando um cálculo saiu do caminho e explorar excitações mistas que envolvem vários orbitais ao mesmo tempo. Os autores também mostram que COOX pode ser estendido de forma natural para tratar moléculas inseridas em sistemas maiores, como um pequeno hóspede dentro de uma gaiola de fulereno ou um cromóforo dentro de um ambiente semelhante a proteína. Nesses casos, o estado excitado é primeiramente definido na molécula de interesse e então incorporado de maneira contínua à descrição quântica completa do entorno, evitando o tedioso gerenciamento manual de orbitais que o ΔSCF frequentemente requer.
O que isso significa para simulações futuras
O estudo conclui que o método ΔCOOX baseado em restrições é pelo menos tão preciso quanto os esquemas ΔSCF estabelecidos e muitas vezes claramente superior em termos de estabilidade numérica e confiabilidade. Para a maioria dos estados excitados que podem ser descritos como um único elétron movendo-se entre orbitais, o ΔCOOX surge como um forte candidato a ferramenta padrão, além de poder servir como um ponto de partida inteligente que ajuda algoritmos ΔSCF mais antigos a convergir. De forma mais ampla, a estrutura completa COOX abre a porta para tratar rotineiramente excitações complicadas e ambientes químicos realistas, aproximando simulações de estado excitado dos sistemas desordenados e diversos encontrados na fotquímica e na ciência de materiais do mundo real.
Citação: Lemke, Y., Kussmann, J. & Ochsenfeld, C. A detailed comparison of ΔSCF methods with the constraint-based orbital-optimized excited state method. Commun Chem 9, 162 (2026). https://doi.org/10.1038/s42004-026-02003-9
Palavras-chave: estados excitados, teoria do funcional da densidade, estrutura eletrônica, transferência de carga, química computacional