Clear Sky Science · tr
CLDN16/CLDN19 mutasyonlarının neden olduğu ailesel hipomagnezemi, hiperkalsiüri ve nefrokalsinozis: dört Çinli ailede olgular
Böbreklerde Minerallerin Dengesi Bozulduğunda
Böbreklerimiz yaşamımızın her dakikasında magnezyum ve kalsiyum gibi minerallerin dengesini sessizce sağlar. Bu süreç aksadığında çocuklarda böbrek taşları, doku skarı ve hatta böbrek yetmezliği gelişebilir. Bu çalışma, mineral dengesini bozan nadir kalıtsal bir böbrek hastalığını paylaşan dört Çinli aileden beş kızı izliyor. Özenli klinik gözlemle modern genetik testleri eşleştirerek araştırmacılar bilinen ve yeni genetik kusurları ortaya çıkardı; bu da doktorların bu sıra dışı hastalığı tanımasını ve yönetmesini kolaylaştırıyor.
Nadir Bir Ailevi Böbrek Durumu
Burada incelenen hastalık ailesel hipomagnezemi ile hiperkalsiüri ve nefrokalsinozis olarak adlandırılır; kısaca FHHNC. FHHNC’li çocuklar idrarla aşırı magnezyum ve kalsiyum kaybeder; bu durum kanda düşük magnezyuma, böbreklerde kalsiyum içerikli birikimlere ve zamanla böbrek fonksiyonunun azalmasına yol açar. Birçoğunda aşırı susama ve sık idrara çıkma veya tekrarlayan üriner enfeksiyonlar görülür. Bazı hastalarda aynı hastalık gözleri de etkiler ve yaşamın erken döneminde görme sorunlarına neden olabilir. Hastalık otozomal resesif kalıtılır; yani ilgili gendeki kusurlu kopyalar her iki ebeveynden alındığında çocuk hasta olur.
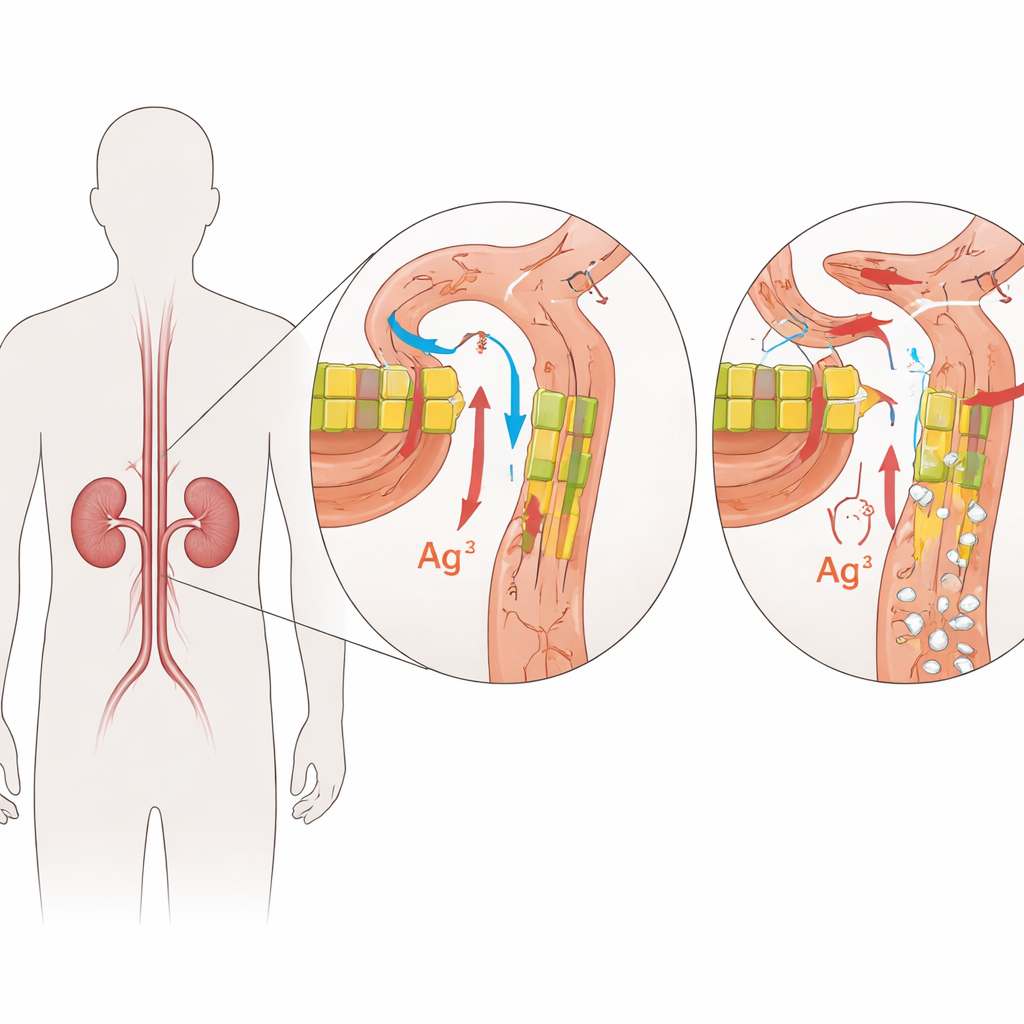
Böbrek Filtrelerinin İçindeki Kapıcılar
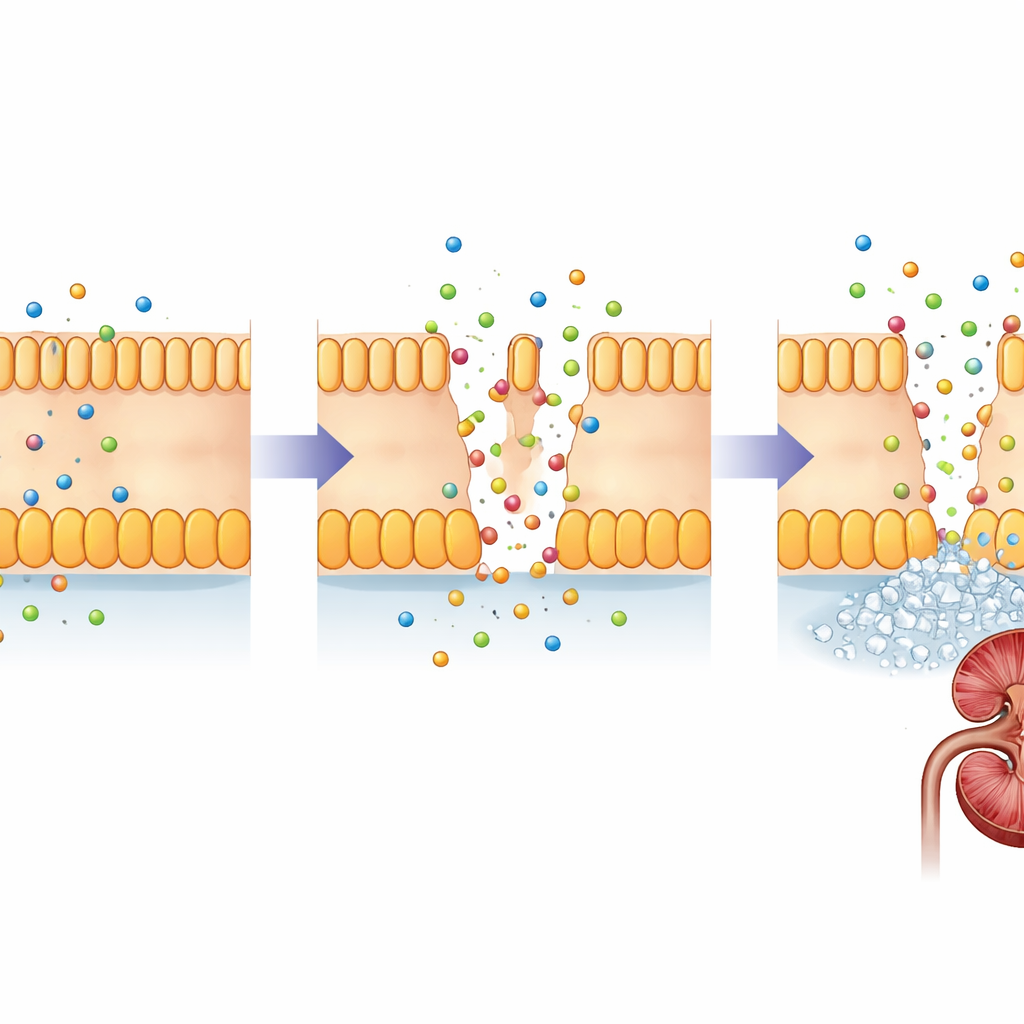
FHHNC, yakından ilişkili iki gen olan CLDN16 ve CLDN19’daki değişikliklerden kaynaklanır. Bu genler, böbrek hücreleri arasındaki dar boşluklarda kapıcı gibi görev yapan küçük proteinler olan claudinleri kodlar. Sağlıklı bir böbrekte claudin-16 ve claudin-19, küçük süzme tüplerinin belirli bir bölümündeki hücre birleşimlerinde bulunur ve magnezyum ile kalsiyumun oluşan idrardan ne kadar geri alındığını ince ayarlar. Bu proteinlerdeki zarar verici değişiklikler bu bariyeri zayıflatır; böylece magnezyum ve kalsiyum kana geri dönmek yerine idrara karışır. Zamanla böbrek dokusundaki fazla kalsiyum taş oluşumunu ve skarı teşvik ederken vücut mineral dengesini korumakta zorlanır.
Aynı Hastalığın Beş Çocuğu, Birçok Yüzü
Araştırma ekibi, genellikle enfeksiyon veya erken ergenlik gibi başka sorunların araştırılması sırasında böbreklerinde kalsiyum birikimi veya taş saptanan, genetik olarak ilişkili olmayan dört Çinli Han ailesinden beş kızı inceledi. Hepsinde kanda düşük magnezyum vardı ve çoğunda kronik böbrek hastalığının erken belirtileri görülüyordu. Ayrıntılı genetik testler, dört kızın CLDN16’da farklı zararlı varyant çiftleri taşıdığını, bir kızın ise daha önce bilinen bir değişiklikle yeni tanımlanan zararlı bir CLDN19 değişikliğinin birleşimini taşıdığını ortaya koydu. Bu varyantların bazıları claudin proteinini kısaltırken; diğerleri yapısını ince şekilde değiştirerek işlevini bozar. Ancak aynı aile içinde bile semptomların şiddeti farklılık gösteriyordu; bu da kalıtsal böbrek hastalığının ne kadar öngörülemez olabileceğini vurguluyor.
Böbreklerin Dışında Beklenmedik İpuçları
Tüm bulgular üriner sistemle sınırlı değildi. CLDN16 değişiklikleri olan bir kızda hemorajik over kisti ve erken ergenlik belirtileri gelişti; bu kombinasyon daha önce bu böbrek hastalığıyla ilişkilendirilmemişti. Claudinler aynı zamanda overler ve gözler dahil olmak üzere diğer organ yüzeylerinin sızdırmazlığını sağlamaya yardımcı olduğundan yazarlar, kusurlu claudin-16 veya claudin-19’un böbrek dışındaki bariyerleri henüz tam olarak anlaşılmamış biçimlerde bozabileceğini öne sürüyor. CLDN19 mutasyonları olan bir diğer kızda ise göz anormallikleri böbrek hastalığı tanınmadan bir yıldan fazla önce saptanmış; bu da görme sorunlarının bu FHHNC formu için erken bir uyarı işareti olabileceğini destekliyor. Birlikte, bu öyküler aynı temel moleküler kusurun çocuklar arasında farklı biçimlerde ortaya çıkabileceğini gösteriyor.
Erken Gen Testinin Önemi
Şu anda kusurlu claudin proteinlerini düzeltebilecek bir tedavi yok, bu nedenle tedavi böbrekleri korumaya odaklanır: bol sıvı alımı, magnezyum ve sitrat takviyeleri ve idrarda kalsiyum kaybını azaltan ilaçlar. Bu önlemlere rağmen birçok hasta sonunda böbrek nakli gerektirebilir. Çin’de bildirilen şimdiye kadarki en büyük çoklu aile FHHNC analizini oluşturan bu çalışma, tıbbi kayda yeni bir CLDN19 varyantı ekliyor ve CLDN16 değişiklikleri olan bir çocukta sıra dışı bir over komplikasyonunu belgeliyor. Aileler ve klinisyenler için mesaj açık: açıklanamayan düşük magnezyum, böbrek taşları veya erken böbrek hasarı olan çocuklarda—özellikle akrabalar etkilendiğinde—genetik test kesin tanı sağlayabilir, böbrek ve göz izlemini yönlendirebilir ve gelecekte ortaya çıkabilecek hedefe yönelik tedavilere kapı aralayabilir.
Atıf: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Anahtar kelimeler: ailevi böbrek hastalığı, magnezyum kaybı, claudin mutasyonları, pediatrik nefrokalsinozis, genetik tübülopatı