Clear Sky Science · de
Familiäre Hypomagnesiämie mit Hyperkalzurie und Nephrokalzinose verursacht durch CLDN16/CLDN19-Mutationen in vier chinesischen Familien
Wenn das Mineralgleichgewicht in den Nieren aus dem Tritt gerät
Unsere Nieren regulieren unbemerkt jede Minute unseres Lebens Mineralien wie Magnesium und Calcium. Wenn dieser Prozess gestört ist, können Kinder Nierensteine, Vernarbungen und sogar Nierenversagen entwickeln. Diese Studie begleitet fünf Mädchen aus vier chinesischen Familien, die eine seltene vererbte Nierenerkrankung teilen, die dieses Mineralgleichgewicht stört. Durch die Kombination aus sorgfältiger klinischer Beobachtung und moderner Gentestung entdeckten die Forschenden sowohl bekannte als auch neue genetische Fehler, was den Ärzten hilft, diese ungewöhnliche Erkrankung besser zu erkennen und zu behandeln.
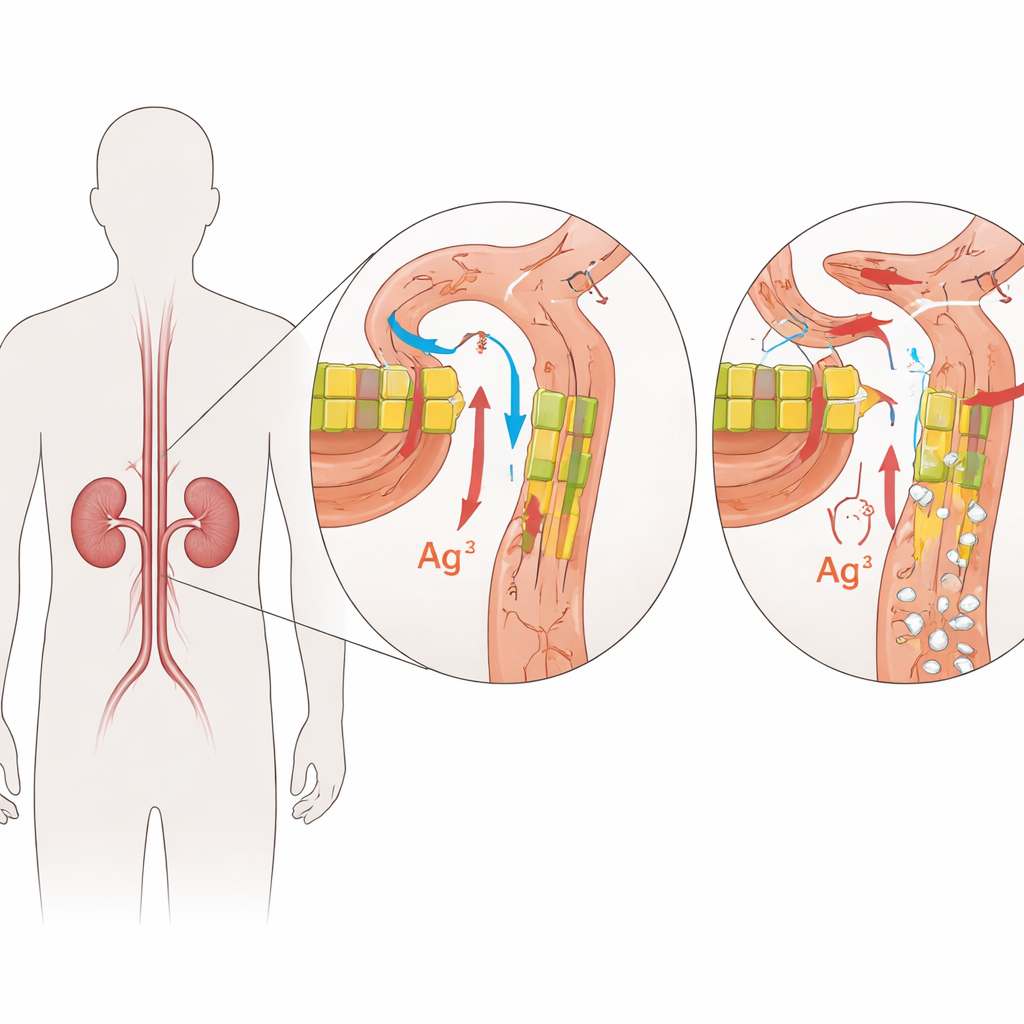
Eine seltene familiäre Nierenerkrankung
Die hier untersuchte Erkrankung heißt familiäre Hypomagnesiämie mit Hyperkalzurie und Nephrokalzinose, kurz FHHNC. Kinder mit FHHNC scheiden zu viel Magnesium und Calcium im Urin aus, was zu niedrigem Blutmagnesium, calciumreichen Ablagerungen in den Nieren und einem allmählichen Funktionsverlust der Nieren führt. Viele trinken und urinieren übermäßig oder leiden an wiederkehrenden Harnwegsinfekten. Bei einigen Patienten betrifft die Erkrankung auch die Augen und verursacht schon früh im Leben Sehstörungen. Die Krankheit wird autosomal-rezessiv vererbt, das heißt, Kinder erkranken erst, wenn sie von beiden Elternteilen eine fehlerhafte Genkopie erhalten.
Die Torwächter in den Nierenfiltern
FHHNC wird durch Veränderungen in zwei eng verwandten Genen verursacht: CLDN16 und CLDN19. Diese Gene kodieren Claudine, kleine Proteine, die wie Torwächter in den engen Zwischenräumen zwischen Nierenzellen wirken. In einer gesunden Niere sitzen Claudin-16 und Claudin-19 in den Verbindungsstellen zwischen Zellen in einem bestimmten Abschnitt der feinen Filterkanälchen und regulieren präzise, wie viel Magnesium und Calcium aus dem sich bildenden Urin zurückgewonnen wird. Schädigende Veränderungen dieser Proteine schwächen diese Barriere, sodass Magnesium und Calcium im Urin verloren gehen, anstatt ins Blut zurückzukehren. Mit der Zeit fördert überschüssiges Calcium im Nierengewebe die Steinbildung und Vernarbung, während der Körper Mühe hat, die Mineralstoffkonzentrationen im Gleichgewicht zu halten.
Fünf Kinder, viele Gesichter derselben Krankheit
Das Forschungsteam untersuchte fünf Mädchen aus vier nicht verwandten chinesischen Han-Familien, bei denen sich Calciumablagerungen oder Steine in den Nieren fanden, häufig im Rahmen von Untersuchungen wegen anderer Probleme wie Infektionen oder vorzeitiger Pubertät. Alle hatten niedriges Blutmagnesium, und die meisten zeigten frühe Anzeichen einer chronischen Nierenerkrankung. Detaillierte genetische Tests ergaben, dass vier Mädchen jeweils verschiedene Paare schädlicher Varianten in CLDN16 trugen, während ein Mädchen eine Kombination aus einer bereits bekannten und einer neu entdeckten schädigenden Veränderung in CLDN19 aufwies. Einige dieser Varianten führen dazu, dass das Claudin-Protein verkürzt wird; andere verändern seine Struktur so, dass es nicht mehr richtig funktioniert. Selbst innerhalb derselben Familie variierten die Symptome jedoch in ihrer Schwere, was die Unvorhersehbarkeit vererbter Nierenerkrankungen unterstreicht.
Unerwartete Hinweise jenseits der Nieren
Nicht alle Befunde beschränkten sich auf den Harntrakt. Ein Mädchen mit CLDN16-Veränderungen entwickelte eine hämorrhagische Ovarialzyste und Anzeichen vorzeitiger Pubertät, eine Kombination, die zuvor nicht mit dieser Nierenerkrankung in Verbindung gebracht wurde. Da Claudine auch dazu beitragen, Oberflächen in anderen Organen wie Ovarien und Augen abzudichten, vermuten die Autorinnen und Autoren, dass fehlerhaftes Claudin-16 oder Claudin-19 Barrieren außerhalb der Niere stören könnte, in einer Weise, die noch nicht vollständig verstanden ist. Ein weiteres Mädchen mit CLDN19-Mutationen wies Augenauffälligkeiten auf, die mehr als ein Jahr vor der Erkennung ihrer Nierenerkrankung festgestellt wurden, was frühere Berichte bestätigt, dass Sehstörungen ein frühes Warnzeichen für diese Form der FHHNC sein können. Zusammen zeigen diese Fälle, dass derselbe grundlegende molekulare Defekt sich bei Kindern sehr unterschiedlich äußern kann.
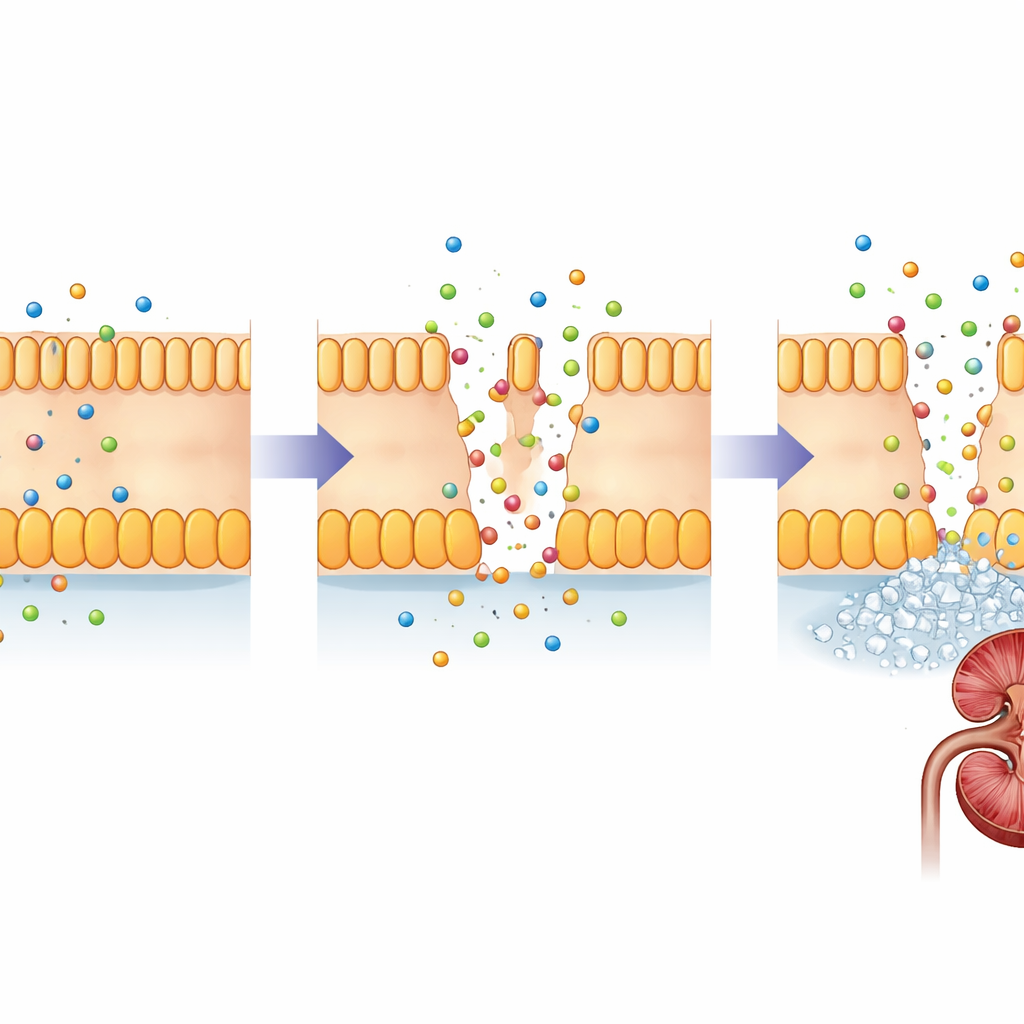
Warum frühe Gentests wichtig sind
Derzeit gibt es keine Heilung, die die fehlerhaften Claudin-Proteine reparieren kann, daher zielt die Behandlung darauf ab, die Nieren zu schützen: reichliche Flüssigkeitszufuhr, Magnesium- und Citratsupplemente sowie Medikamente, die den Calciumausscheidungsverlust im Urin verringern. Selbst bei solcher Versorgung benötigen viele Patientinnen und Patienten letztlich eine Nierentransplantation. Diese Studie, die bislang größte Mehrfamilienanalyse von FHHNC in China, ergänzt die medizinische Datenbank um eine neue CLDN19-Variante und dokumentiert eine ungewöhnliche ovarielle Komplikation bei einem Kind mit CLDN16-Veränderungen. Für Familien und Kliniker ist die Botschaft klar: Bei Kindern mit unerklärlich niedrigem Magnesium, Nierensteinen oder frühem Nierenschaden — besonders wenn Angehörige betroffen sind — kann eine genetische Untersuchung eine gesicherte Diagnose liefern, die Überwachung von Nieren und Augen lenken und den Weg für künftig verfügbare gezielte Therapien öffnen.
Zitation: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Schlüsselwörter: familiäre Nierenerkrankung, Magnesiumverlust, Claudin-Mutationen, pädiatrische Nephrokalzinose, genetische Tubulopathie