Clear Sky Science · pl
Rodzinna hipomagnezemia z hiperkalciurią i nefrokalcynozą spowodowana mutacjami CLDN16/CLDN19 w czterech chińskich rodzinach
Gdy równowaga minerałów w nerkach zawodzi
Nasze nerki nieprzerwanie regulują poziomy minerałów, takich jak magnez i wapń. Gdy ten proces się zakłóca, u dzieci mogą pojawić się kamienie nerkowe, bliznowacenie i nawet niewydolność nerek. W badaniu opisano pięć dziewczynek z czterech chińskich rodzin, które mają rzadkie, dziedziczne schorzenie zaburzające tę równowagę mineralną. Połączenie uważnej obserwacji klinicznej z nowoczesnymi badaniami genetycznymi pozwoliło badaczom odnaleźć zarówno znane, jak i nowe mutacje, co pomaga lekarzom lepiej rozpoznawać i leczyć tę nietypową chorobę.
Rzadkie rodzinne schorzenie nerek
Opisywane schorzenie nosi nazwę rodzinna hipomagnezemia z hiperkalciurią i nefrokalcynozą, w skrócie FHHNC. Dzieci z FHHNC tracą za dużo magnezu i wapnia z moczem, co prowadzi do niskiego poziomu magnezu we krwi, odkładania się złogów wapniowych w nerkach i stopniowego pogarszania funkcji nerek. Wielu pacjentów ma też nadmierne pragnienie i oddawanie moczu lub nawracające infekcje układu moczowego. U niektórych chorych choroba dotyka też oczu, powodując problemy ze wzrokiem we wczesnym okresie życia. Schorzenie dziedziczy się autosomalnie recesywnie, co oznacza, że choroba ujawnia się, gdy dziecko otrzyma wadliwą kopię genu od obojga rodziców.
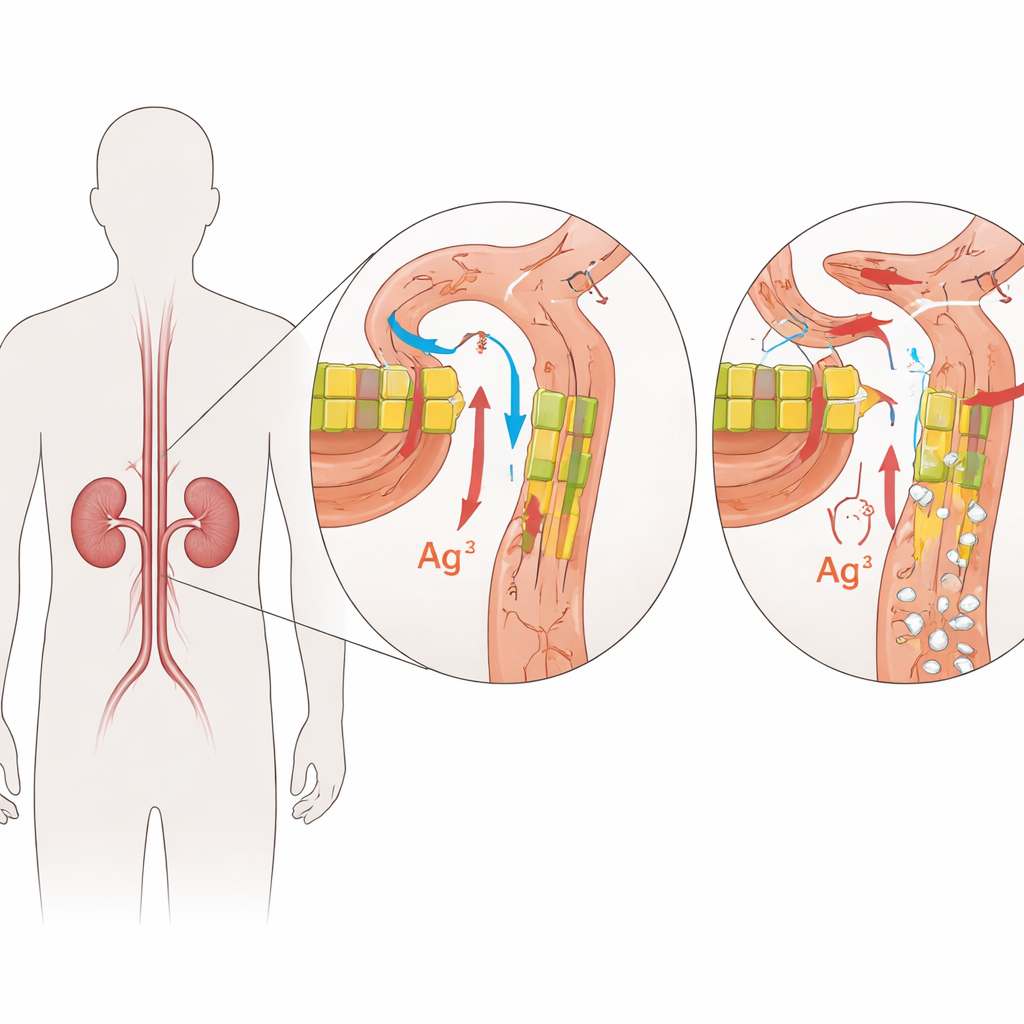
Stróże w szczelinach filtrów nerkowych
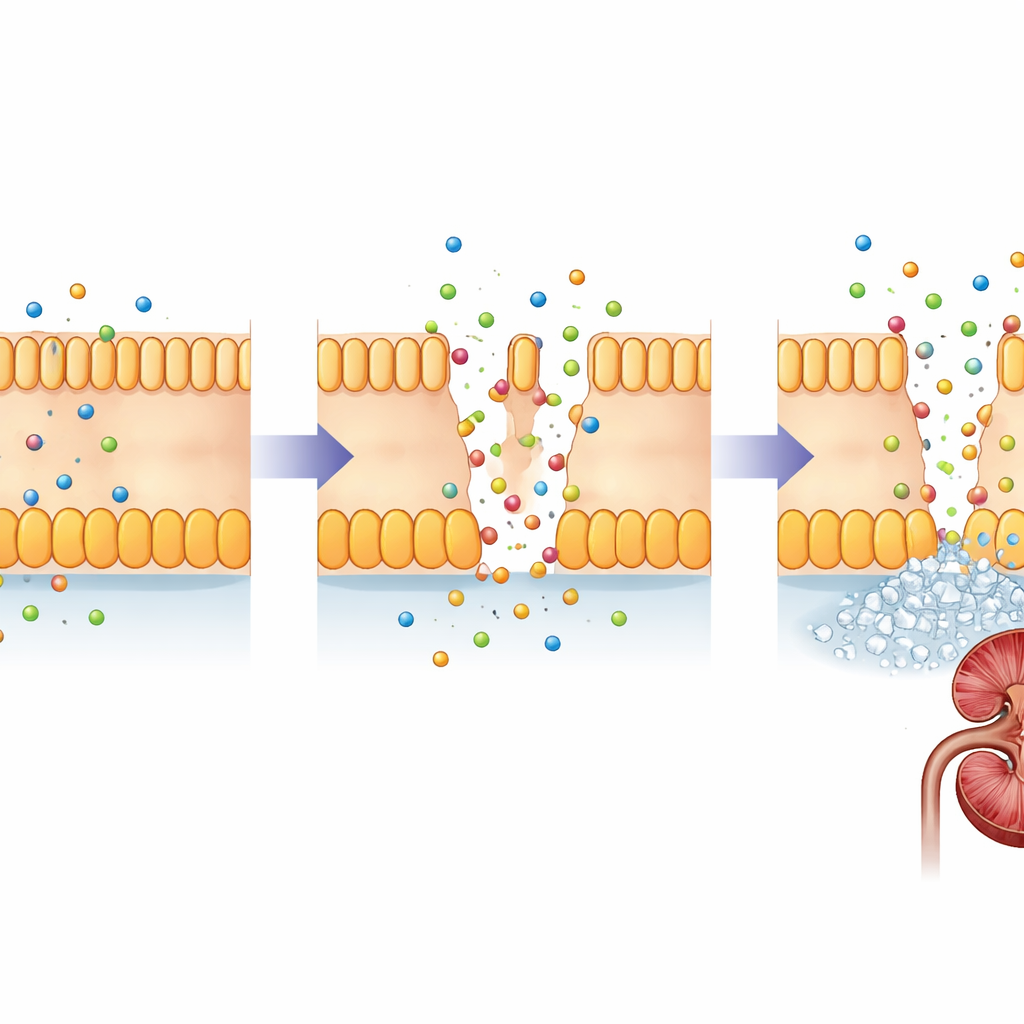
FHHNC spowodowane jest zmianami w dwóch blisko spokrewnionych genach, CLDN16 i CLDN19. Geny te kodują klaudyny, niewielkie białka pełniące rolę strażników w wąskich przestrzeniach między komórkami nerek. W zdrowej nerce klaudyna‑16 i klaudyna‑19 lokalizują się w połączeniach międzykomórkowych w określonym odcinku kanalików nerkowych, gdzie precyzyjnie regulują, ile magnezu i wapnia jest odzyskiwane z tworzącego się moczu. Uszkadzające zmiany w tych białkach osłabiają barierę, przez co magnez i wapń „uciekają” z moczem zamiast wracać do krwi. Z czasem nadmiar wapnia w tkance nerkowej sprzyja tworzeniu kamieni i bliznowaceniu, a organizm ma trudności z utrzymaniem równowagi mineralnej.
Pięć dzieci, wiele twarzy tej samej choroby
Zespół badawczy przeanalizował pięć dziewczynek z czterech niespokrewnionych chińskich rodzin Han, u których stwierdzono złogi wapnia lub kamienie w nerkach, często podczas badań z powodu innych problemów, takich jak infekcje czy przedwczesne dojrzewanie. Wszystkie miały niski poziom magnezu we krwi, a większość wykazywała wczesne oznaki przewlekłej choroby nerek. Szczegółowe badania genetyczne wykazały, że u czterech dziewczynek znaleziono różne pary szkodliwych wariantów w CLDN16, natomiast u jednej dziewczynki zidentyfikowano kombinację wcześniej znanej i nowo odkrytej zmiany w CLDN19. Niektóre z tych wariantów powodują skrócenie białka klaudyny; inne subtelnie zmieniają jego strukturę, uniemożliwiając prawidłowe funkcjonowanie. Nawet w obrębie tej samej rodziny nasilenie objawów bywało różne, co podkreśla nieprzewidywalność dziedzicznych chorób nerek.
Niespodziewane wskazówki poza nerkami
Nie wszystkie obserwacje ograniczały się do układu moczowego. Jedna dziewczynka z mutacjami w CLDN16 rozwinęła krwotoczny torbiel jajnika i objawy przedwczesnego dojrzewania — kombinację wcześniej niepowiązaną z tym schorzeniem nerek. Ponieważ klaudyny uczestniczą także w uszczelnianiu powierzchni innych narządów, w tym jajników i oczu, autorzy sugerują, że wadliwa klaudyna‑16 lub klaudyna‑19 może zaburzać bariery poza nerką w sposób jeszcze nie w pełni poznany. Inna dziewczynka z mutacjami w CLDN19 miała nieprawidłowości okulistyczne wykryte ponad rok przed rozpoznaniem choroby nerek, co potwierdza wcześniejsze doniesienia, że problemy ze wzrokiem mogą być wczesnym znakiem tej postaci FHHNC. Te przypadki pokazują, że ten sam podstawowy defekt molekularny może przejawiać się u każdego dziecka inaczej.
Dlaczego wczesne badania genetyczne są ważne
Obecnie nie ma leczenia, które naprawiłoby wadliwe białka klaudynowe, dlatego terapia koncentruje się na ochronie nerek: odpowiednim nawodnieniu, suplementacji magnezem i cytrynianami oraz lekach zmniejszających utratę wapnia z moczem. Nawet przy takim postępowaniu wielu pacjentów ostatecznie wymaga transplantacji nerki. To badanie, największa do tej pory wielorodzinna analiza FHHNC w Chinach, dodaje do rejestru medycznego nowy wariant CLDN19 i dokumentuje nietypowe powikłanie jajnikowe u dziecka z mutacjami CLDN16. Dla rodzin i lekarzy przesłanie jest jasne: u dzieci z niewyjaśnioną hipomagnezemią, kamicą nerkową lub wczesnym uszkodzeniem nerek — zwłaszcza gdy występują przypadki w rodzinie — badania genetyczne mogą dać pewne rozpoznanie, ukierunkować monitorowanie nerek i oczu oraz otworzyć drogę do nowych celowanych terapii w przyszłości.
Cytowanie: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Słowa kluczowe: rodzinna choroba nerek, utrata magnezu, mutacje klaudyn, pediatryczna nefrokalcynoza, genetyczna kanalikopatia