Clear Sky Science · it
Ipomagnesemia familiare con ipercalciuria e nefrocalcinosi dovuta a mutazioni CLDN16/CLDN19 in quattro famiglie cinesi
Quando l’equilibrio dei minerali si rompe nei reni
I nostri reni bilanciano silenziosamente minerali come magnesio e calcio ogni minuto della nostra vita. Quando questo processo viene compromesso, i bambini possono sviluppare calcoli renali, cicatrizzazione e persino insufficienza renale. Questo studio segue cinque bambine provenienti da quattro famiglie cinesi che condividono un raro disturbo renale ereditario che altera questo equilibrio minerale. Abbinando un’attenta osservazione clinica a moderne analisi genetiche, i ricercatori hanno scoperto sia anomalie genetiche note sia nuove varianti, aiutando i medici a riconoscere e gestire meglio questa malattia insolita.
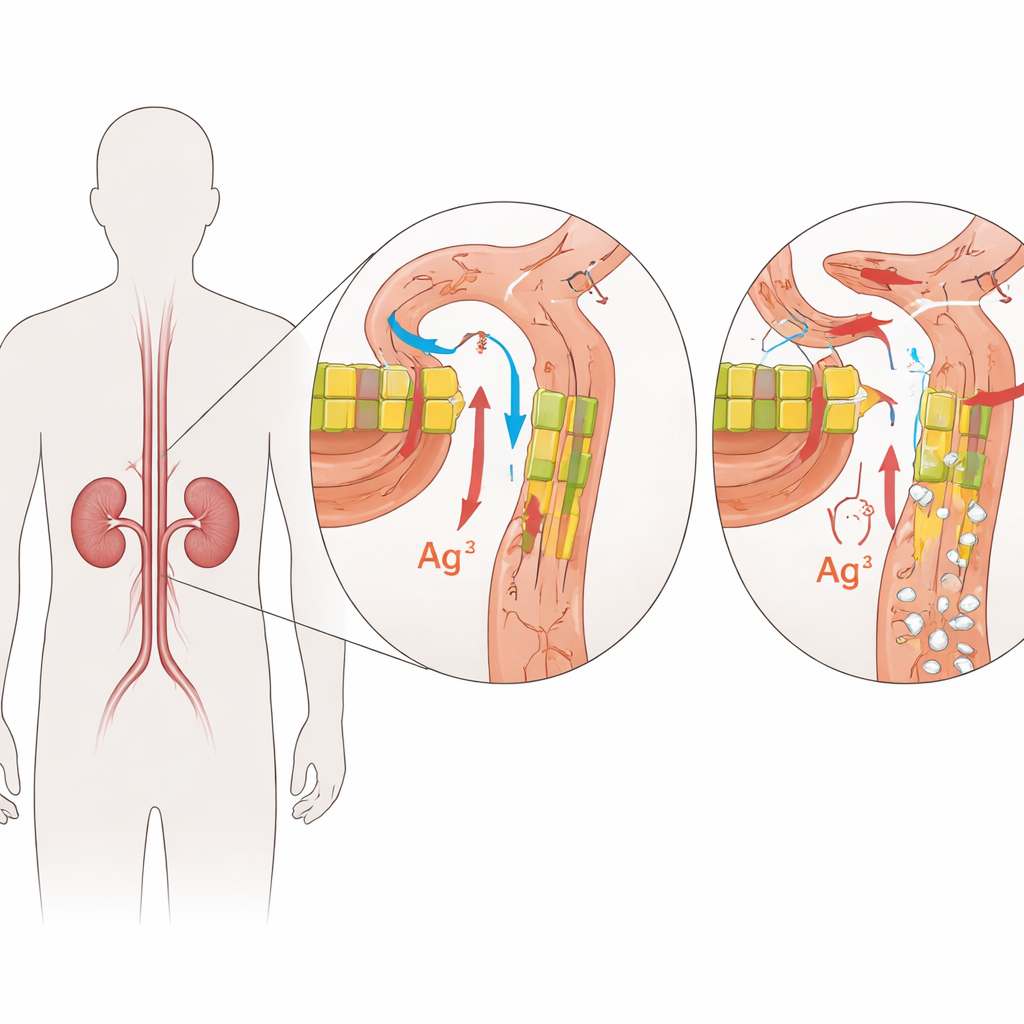
Una rara condizione renale famigliare
Il disturbo esaminato qui è chiamato ipomagnesemia familiare con ipercalciuria e nefrocalcinosi, o FHHNC. I bambini con FHHNC perdono troppo magnesio e calcio nelle urine, il che porta a bassi livelli di magnesio nel sangue, depositi ricchi di calcio nei reni e progressiva perdita della funzione renale. Molti presentano anche sete e minzione eccessive o infezioni urinarie ricorrenti. In alcuni pazienti, la stessa condizione interessa anche gli occhi, causando problemi di vista precoci nella vita. La malattia si eredita con un modello autosomico recessivo, il che significa che i bambini si ammalano solo quando ricevono una copia difettosa del gene rilevante da entrambi i genitori.
I guardiani nei filtri renali
La FHHNC è causata da alterazioni in due geni strettamente correlati, CLDN16 e CLDN19. Questi geni codificano le claudine, piccole proteine che agiscono come guardiani negli stretti spazi tra le cellule renali. In un rene sano, claudina-16 e claudina-19 si trovano nelle giunzioni tra le cellule in un segmento specifico dei sottili tubuli filtranti, dove regolano con precisione quanto magnesio e calcio vengono recuperati dall’urina in formazione. Le variazioni dannose in queste proteine indeboliscono questa barriera, per cui magnesio e calcio sfuggono nelle urine invece di essere restituiti al flusso sanguigno. Nel tempo, l’eccesso di calcio nel tessuto renale favorisce la formazione di calcoli e la cicatrizzazione, mentre l’organismo fatica a mantenere l’equilibrio dei minerali.
Cinque bambine, molti volti della stessa malattia
Il team di ricerca ha studiato cinque bambine provenienti da quattro famiglie Han cinesi non correlate, nelle quali sono stati trovati depositi di calcio o calcoli nei reni, spesso durante esami per altri problemi come infezioni o pubertà precoce. Tutte avevano bassi livelli di magnesio nel sangue e la maggior parte mostrava segni precoci di malattia renale cronica. L’analisi genetica dettagliata ha rivelato che quattro bambine portavano diverse coppie di varianti dannose in CLDN16, mentre una bambina presentava una combinazione di una variazione nota e di una nuova variazione dannosa in CLDN19. Alcune di queste varianti causano l’abbreviazione della proteina claudina; altre ne alterano sottilmente la struttura impedendone il corretto funzionamento. Tuttavia, anche all’interno della stessa famiglia, i sintomi variavano in gravità, evidenziando quanto sia imprevedibile la malattia renale ereditaria.
Indizi inaspettati oltre i reni
Non tutte le osservazioni si sono limitate alle vie urinarie. Una bambina con variazioni in CLDN16 ha sviluppato una cisti ovarica emorragica e segni di pubertà precoce, una combinazione non collegata in precedenza a questo disturbo renale. Poiché le claudine contribuiscono anche a sigillare le superfici in altri organi, comprese ovaie e occhi, gli autori suggeriscono che una claudina-16 o claudina-19 difettosa potrebbe alterare barriere oltre il rene in modi non ancora pienamente compresi. Un’altra bambina con mutazioni in CLDN19 aveva anomalie oculari rilevate più di un anno prima che la sua malattia renale fosse riconosciuta, richiamando segnalazioni precedenti secondo cui i problemi visivi possono essere un campanello d’allarme precoce per questa forma di FHHNC. Nel loro insieme, queste storie mostrano che lo stesso difetto molecolare di base può manifestarsi in modi diversi da un bambino all’altro.
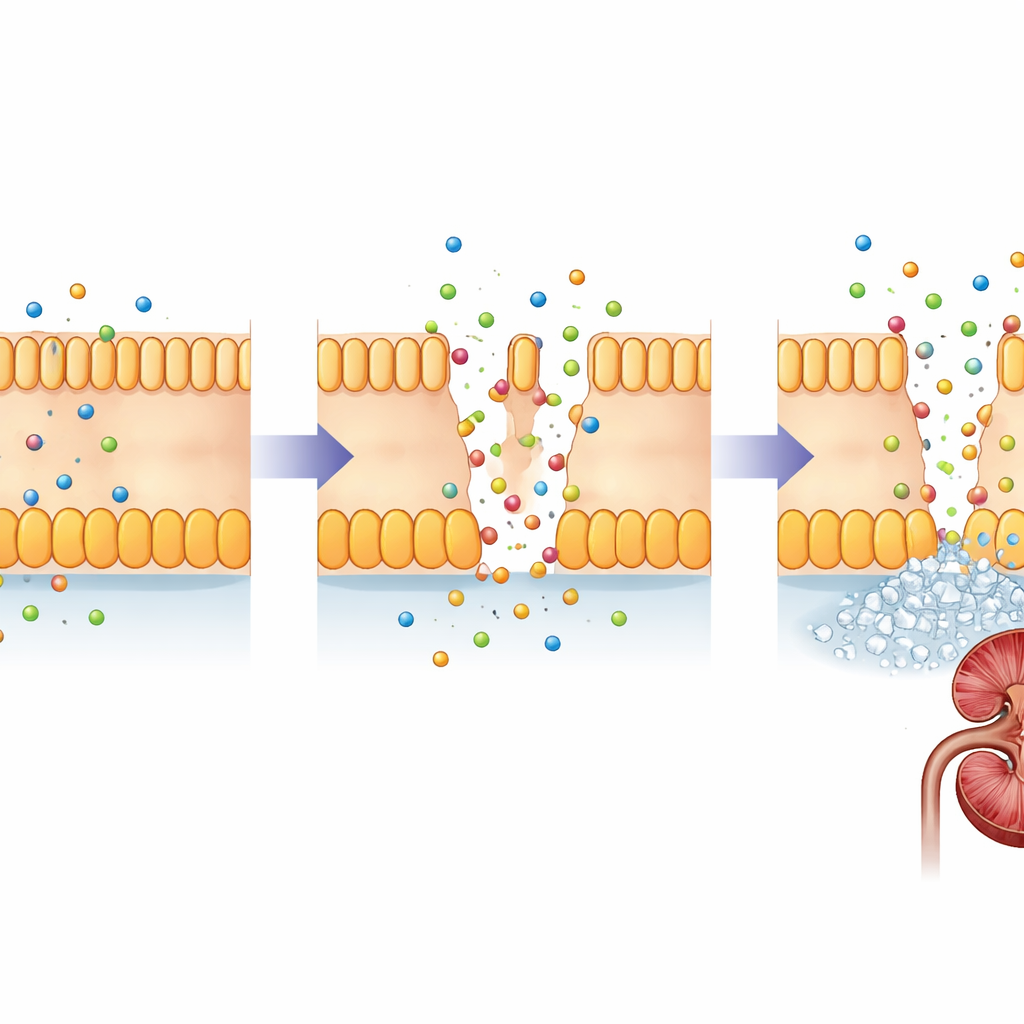
Perché è importante il test genetico precoce
Al momento non esiste una cura in grado di riparare le proteine claudine difettose, quindi il trattamento si concentra sulla protezione dei reni: un’adeguata assunzione di liquidi, integrazioni di magnesio e citrati e farmaci che riducono la perdita di calcio nelle urine. Anche con tali cure, molti pazienti finiscono per necessitare un trapianto renale. Questo studio, la più ampia analisi multi‑famiglia di FHHNC finora riportata in Cina, aggiunge una nuova variante di CLDN19 alla letteratura medica e documenta una insolita complicanza ovarica in una bambina con alterazioni di CLDN16. Per famiglie e clinici, il messaggio è chiaro: nei bambini con ipomagnesemia inspiegata, calcoli renali o danno renale precoce—soprattutto quando sono presenti parenti affetti—il test genetico può fornire una diagnosi definitiva, guidare il monitoraggio di reni e occhi e aprire la strada a terapie mirate emergenti in futuro.
Citazione: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Parole chiave: malattia renale familiare, perdita di magnesio, mutazioni delle claudine, nefrocalcinosi pediatrica, tubulopatia genetica