Clear Sky Science · en
Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families
When Mineral Balance Goes Wrong in the Kidneys
Our kidneys quietly balance minerals like magnesium and calcium every minute of our lives. When this process falters, children can develop kidney stones, scarring, and even kidney failure. This study follows five girls from four Chinese families who share a rare inherited kidney disorder that upsets this mineral balance. By pairing careful clinical observation with modern gene testing, the researchers uncovered both known and new genetic glitches, helping doctors better recognize and manage this unusual disease.
A Rare Family Kidney Condition
The disorder examined here is called familial hypomagnesemia with hypercalciuria and nephrocalcinosis, or FHHNC for short. Children with FHHNC lose too much magnesium and calcium in their urine, which leads to low blood magnesium, calcium-rich deposits in the kidneys, and gradual loss of kidney function. Many also drink and urinate excessively or suffer repeated urinary infections. In some patients, the same condition also affects the eyes, causing vision problems early in life. The illness is inherited in an autosomal recessive pattern, meaning children become sick only when they receive a faulty copy of the relevant gene from both parents.
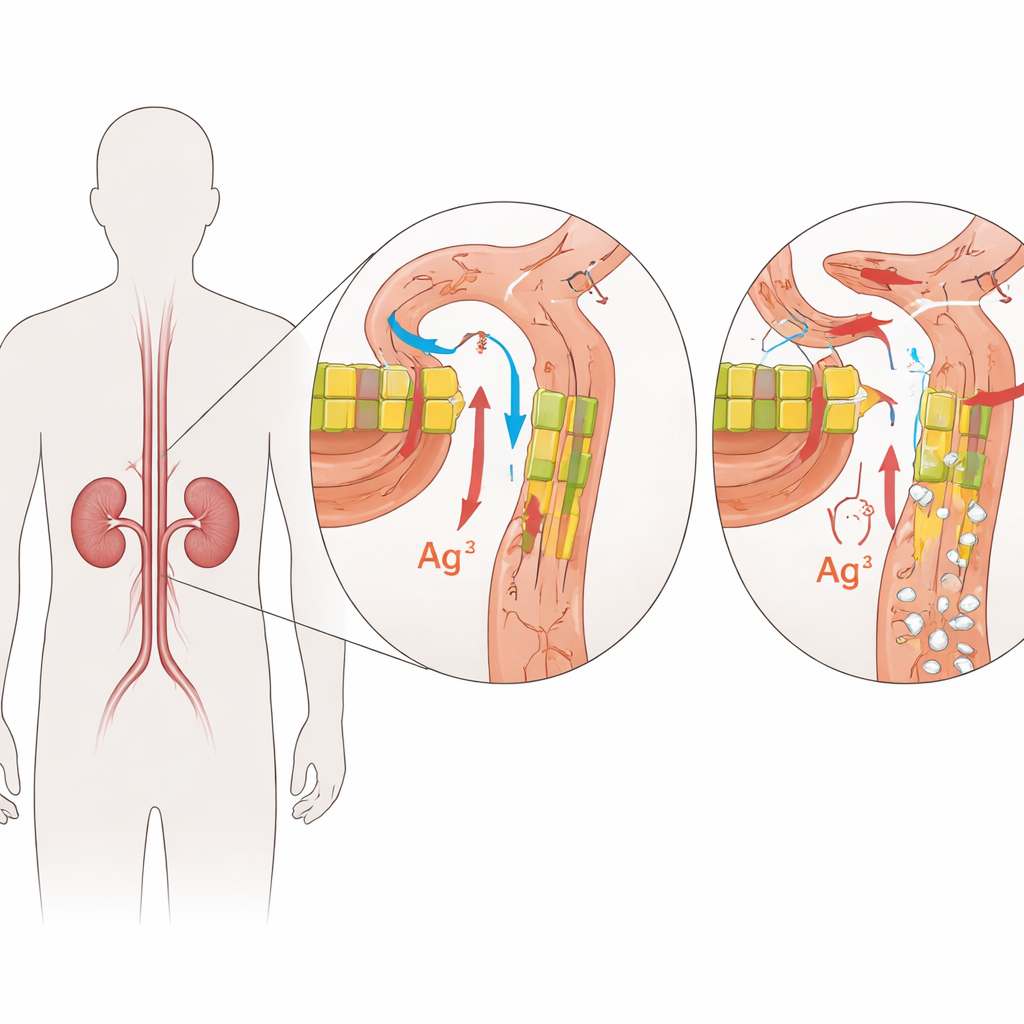
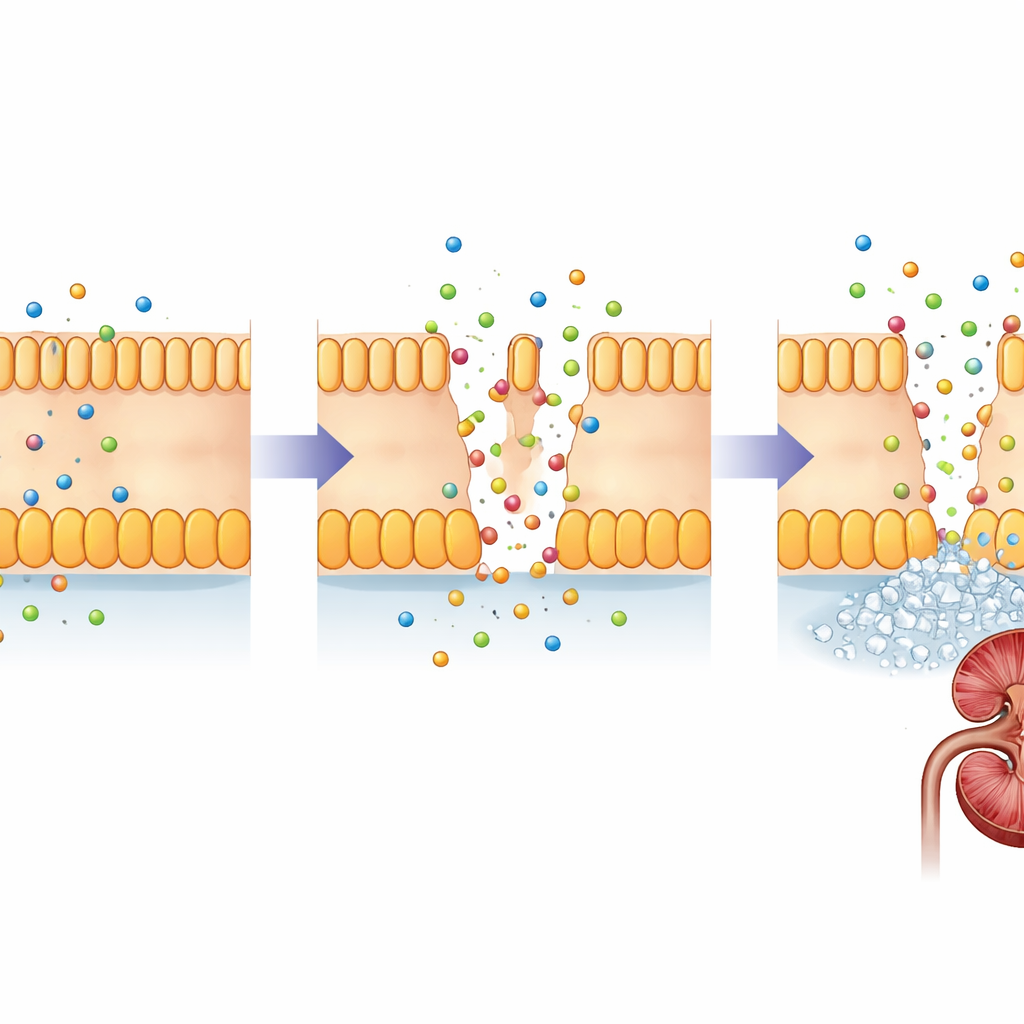
The Gatekeepers Inside Kidney Filters
FHHNC is caused by changes in two closely related genes, CLDN16 and CLDN19. These genes encode claudins, small proteins that act like gatekeepers in the narrow spaces between kidney cells. In a healthy kidney, claudin-16 and claudin-19 sit in the junctions between cells in a specific segment of the tiny filtering tubes, where they fine‑tune how much magnesium and calcium are reclaimed from the forming urine. Damaging changes in these proteins weaken this barrier, so magnesium and calcium slip away into the urine instead of being returned to the bloodstream. Over time, excess calcium in the kidney tissue promotes stone formation and scarring, while the body struggles to keep mineral levels in balance.
Five Children, Many Faces of the Same Disease
The research team studied five girls from four unrelated Chinese Han families who were found to have calcium deposits or stones in their kidneys, often during tests for other problems such as infections or early puberty. All had low blood magnesium, and most showed early signs of chronic kidney disease. Detailed genetic testing revealed that four girls carried different pairs of harmful variants in CLDN16, while one girl carried a combination of a previously known and a newly discovered damaging change in CLDN19. Some of these variants cause the claudin protein to be cut short; others subtly alter its structure so that it no longer functions properly. Even within the same family, however, symptoms varied in severity, highlighting how unpredictable inherited kidney disease can be.
Unexpected Clues Beyond the Kidneys
Not all the findings were confined to the urinary tract. One girl with CLDN16 changes developed a hemorrhagic ovarian cyst and signs of early puberty, a combination not previously linked to this kidney disorder. Because claudins also help seal surfaces in other organs, including the ovaries and eyes, the authors suggest that faulty claudin-16 or claudin-19 might disrupt barriers beyond the kidney in ways that are not yet fully understood. Another girl with CLDN19 mutations had eye abnormalities detected more than a year before her kidney disease was recognized, echoing earlier reports that vision problems can be an early warning sign for this form of FHHNC. Together, these stories show that the same basic molecular defect can manifest in diverse ways from child to child.
Why Early Gene Testing Matters
At present, there is no cure that can fix the faulty claudin proteins, so treatment focuses on protecting the kidneys: generous fluid intake, magnesium and citrate supplements, and medications that reduce calcium loss in the urine. Even with such care, many patients eventually require kidney transplantation. This study, the largest multi‑family analysis of FHHNC reported so far in China, adds a new CLDN19 variant to the medical record and documents an unusual ovarian complication in a child with CLDN16 changes. For families and clinicians, the message is clear: in children with unexplained low magnesium, kidney stones, or early kidney damage—especially when relatives are affected—genetic testing can provide a firm diagnosis, guide monitoring of kidneys and eyes, and open the door to emerging targeted treatments in the future.
Citation: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Keywords: familial kidney disease, magnesium wasting, claudin mutations, pediatric nephrocalcinosis, genetic tubulopathy