Clear Sky Science · pt
Hipomagnesemia familiar com hipercalciúria e nefrocalcinose causada por mutações em CLDN16/CLDN19 em quatro famílias chinesas
Quando o equilíbrio mineral falha nos rins
Nossos rins equilibram silenciosamente minerais como magnésio e cálcio a cada minuto de nossas vidas. Quando esse processo falha, crianças podem desenvolver cálculos renais, cicatrização e até insuficiência renal. Este estudo acompanha cinco meninas de quatro famílias chinesas que compartilham uma rara doença renal hereditária que perturba esse equilíbrio mineral. Ao combinar observação clínica cuidadosa com testes genéticos modernos, os pesquisadores identificaram variantes genéticas conhecidas e novas, ajudando médicos a reconhecer e manejar melhor essa doença incomum.
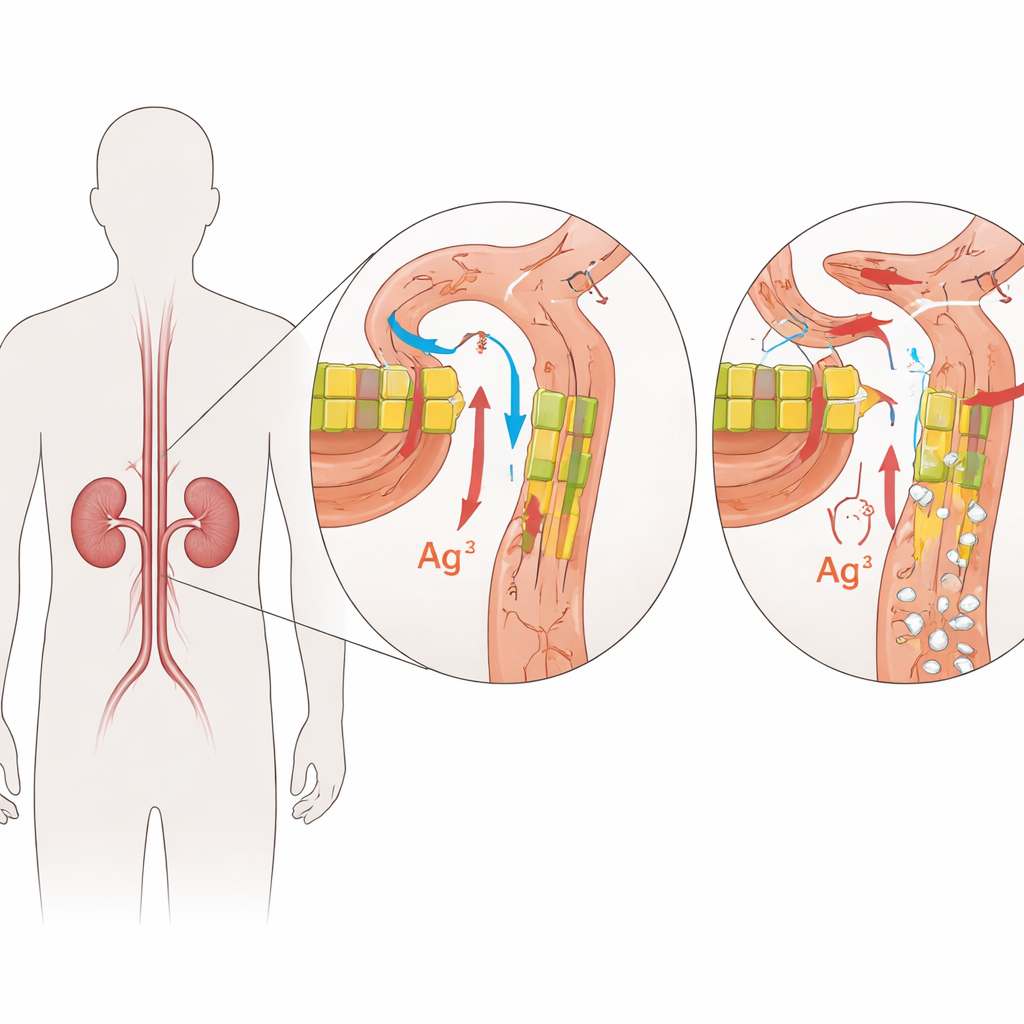
Uma condição renal familiar rara
A desordem examinada aqui chama‑se hipomagnesemia familiar com hipercalciúria e nefrocalcinose, ou FHHNC. Crianças com FHHNC perdem magnésio e cálcio demais na urina, o que leva a baixos níveis sanguíneos de magnésio, depósitos ricos em cálcio nos rins e perda gradual da função renal. Muitas também bebem e urinam em excesso ou sofrem infecções urinárias recorrentes. Em alguns pacientes, a mesma condição afeta os olhos, causando problemas de visão precocemente. A doença é herdada de forma autossômica recessiva, o que significa que as crianças adoecem somente quando recebem uma cópia defeituosa do gene relevante de ambos os pais.
Os porteiros dentro dos filtros renais
A FHHNC é causada por alterações em dois genes intimamente relacionados, CLDN16 e CLDN19. Esses genes codificam as claudinas, pequenas proteínas que atuam como porteiros nos espaços estreitos entre as células renais. Em um rim saudável, claudina‑16 e claudina‑19 ocupam as junções entre células em um segmento específico dos minúsculos túbulos filtrantes, onde ajustam quanto magnésio e cálcio são recuperados da urina em formação. Alterações deletérias nessas proteínas enfraquecem essa barreira, de modo que magnésio e cálcio escapam pela urina em vez de retornarem à corrente sanguínea. Com o tempo, o excesso de cálcio no tecido renal favorece a formação de cálculos e cicatrização, enquanto o corpo luta para manter o equilíbrio mineral.
Cinco crianças, muitas faces da mesma doença
A equipe estudou cinco meninas de quatro famílias Han chinesas não aparentadas que foram encontradas com depósitos de cálcio ou cálculos nos rins, frequentemente durante exames para outros problemas, como infecções ou puberdade precoce. Todas apresentavam baixo magnésio no sangue, e a maioria mostrou sinais iniciais de doença renal crônica. Testes genéticos detalhados revelaram que quatro meninas carregavam pares diferentes de variantes prejudiciais em CLDN16, enquanto uma menina apresentava uma combinação de uma alteração previamente conhecida e uma nova alteração deletéria em CLDN19. Algumas dessas variantes resultam em uma proteína claudina encurtada; outras alteram sutilmente sua estrutura de modo que ela não funcione adequadamente. Mesmo dentro da mesma família, entretanto, os sintomas variaram em gravidade, destacando o quão imprevisível a doença renal hereditária pode ser.
Pistas inesperadas além dos rins
Nem todos os achados se limitaram ao trato urinário. Uma menina com alterações em CLDN16 desenvolveu um cisto ovariano hemorrágico e sinais de puberdade precoce, uma combinação não previamente ligada a esse distúrbio renal. Como as claudinas também ajudam a selar superfícies em outros órgãos, incluindo ovários e olhos, os autores sugerem que claudina‑16 ou claudina‑19 defeituosas podem perturbar barreiras além do rim de formas ainda pouco compreendidas. Outra menina com mutações em CLDN19 teve anomalias oculares detectadas mais de um ano antes do reconhecimento da doença renal, ecoando relatos anteriores de que problemas de visão podem ser um sinal precoce desta forma de FHHNC. Juntas, essas histórias mostram que o mesmo defeito molecular básico pode se manifestar de maneiras diversas de criança para criança.
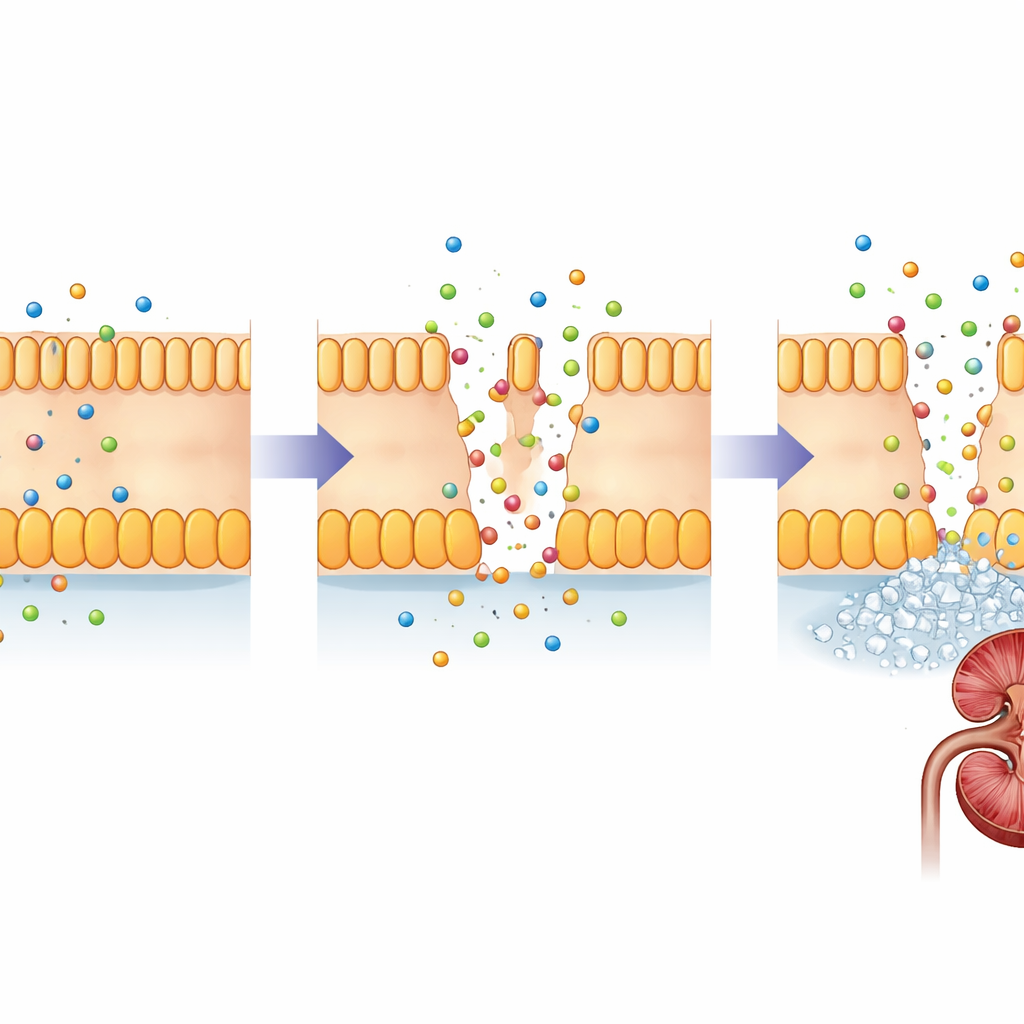
Por que o teste genético precoce é importante
Atualmente, não existe cura que corrija as proteínas claudinas defeituosas, portanto o tratamento concentra‑se em proteger os rins: ingestão adequada de líquidos, suplementação de magnésio e citrato, e medicamentos que reduzem a perda de cálcio pela urina. Mesmo com esses cuidados, muitos pacientes acabam necessitando de transplante renal. Este estudo, a maior análise múltipla de famílias com FHHNC relatada até agora na China, acrescenta uma nova variante em CLDN19 ao registro médico e descreve uma complicação ovariana incomum em uma criança com alterações em CLDN16. Para famílias e clínicos, a mensagem é clara: em crianças com hipomagnesemia inexplicada, cálculos renais ou dano renal precoce—especialmente quando parentes são afetados—o teste genético pode fornecer um diagnóstico firme, orientar o monitoramento de rins e olhos e abrir caminho para tratamentos direcionados emergentes no futuro.
Citação: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Palavras-chave: doença renal familiar, perda de magnésio, mutações em claudinas, nefrocalcinose pediátrica, tubulopatia genética