Clear Sky Science · es
Hipomagnesemia familiar con hipercalciuria y nefrocalcinosis causada por mutaciones en CLDN16/CLDN19 en cuatro familias chinas
Cuando el equilibrio mineral falla en los riñones
Nuestros riñones regulan de forma silenciosa minerales como el magnesio y el calcio cada minuto de nuestra vida. Cuando este proceso falla, los niños pueden desarrollar cálculos renales, fibrosis e incluso insuficiencia renal. Este estudio sigue a cinco niñas de cuatro familias chinas que comparten un raro trastorno renal hereditario que altera este equilibrio mineral. Al combinar una observación clínica cuidadosa con pruebas genéticas modernas, los investigadores identificaron fallos genéticos conocidos y nuevos, ayudando a los médicos a reconocer y manejar mejor esta enfermedad poco común.
Una rara enfermedad renal familiar
El trastorno analizado aquí se denomina hipomagnesemia familiar con hipercalciuria y nefrocalcinosis, o FHHNC por sus siglas en inglés. Los niños con FHHNC pierden demasiado magnesio y calcio en la orina, lo que conduce a niveles bajos de magnesio en sangre, depósitos ricos en calcio en los riñones y una pérdida gradual de la función renal. Muchos también presentan sed y micción excesivas o infecciones urinarias recurrentes. En algunos pacientes, la misma afección afecta también a los ojos, causando problemas de visión a edad temprana. La enfermedad se hereda en un patrón autosómico recesivo, lo que significa que los niños enferman sólo cuando reciben una copia defectuosa del gen relevante de ambos progenitores.
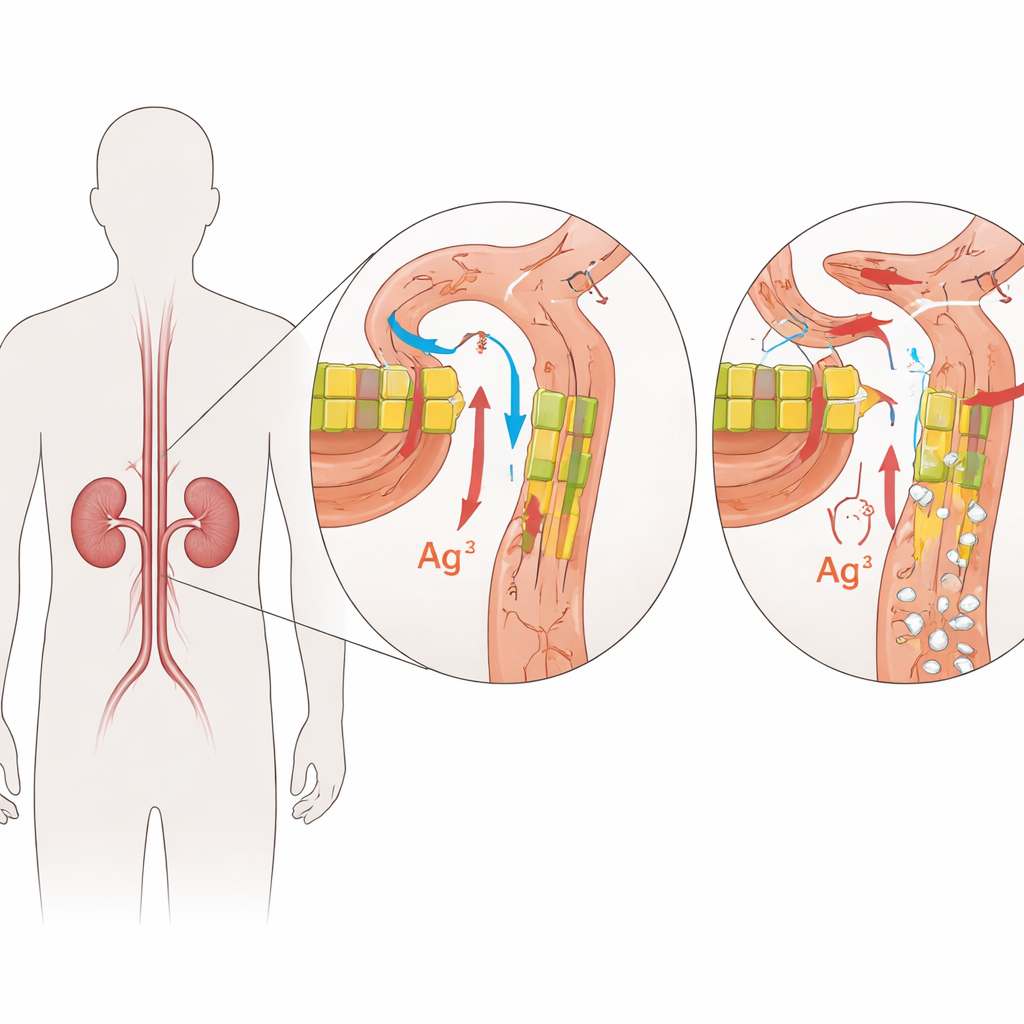
Los guardianes dentro de los filtros renales
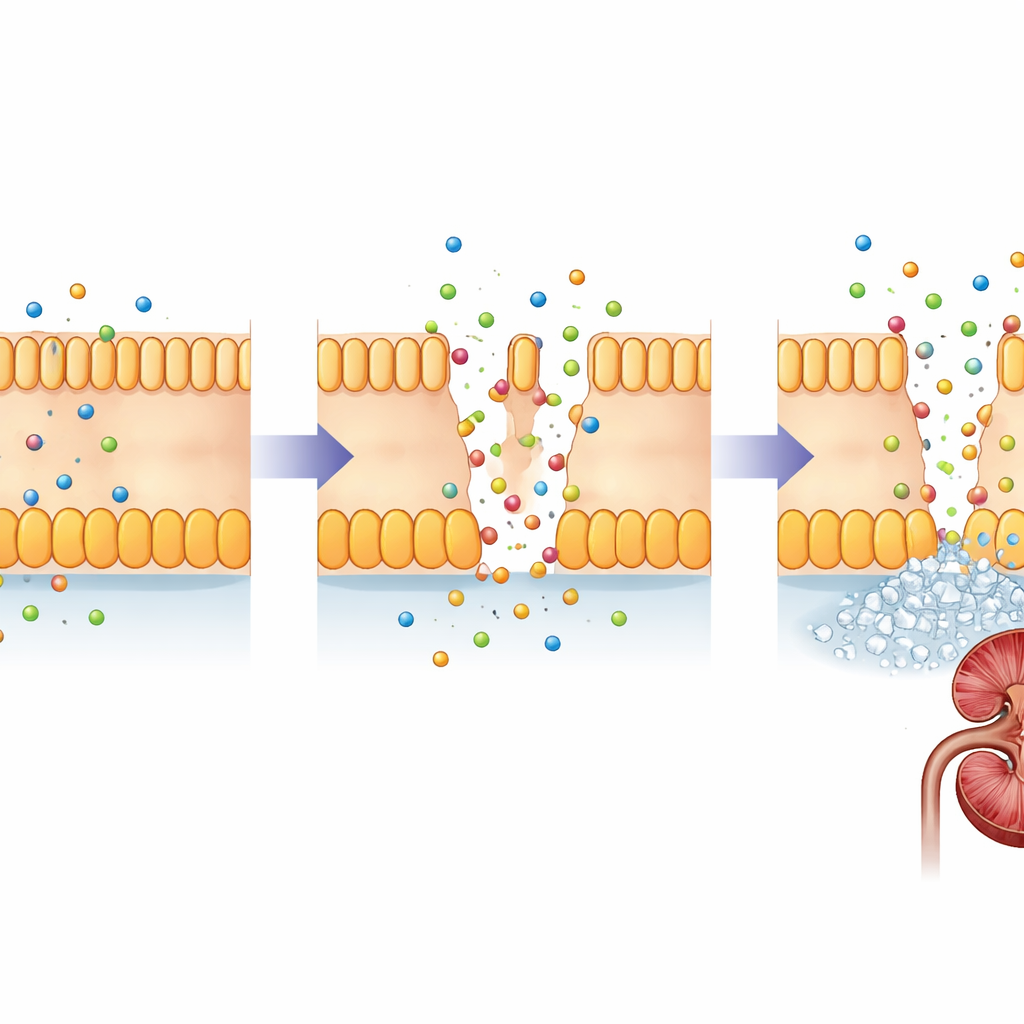
La FHHNC es causada por cambios en dos genes estrechamente relacionados, CLDN16 y CLDN19. Estos genes codifican claudinas, pequeñas proteínas que actúan como guardianes en los estrechos espacios entre las células renales. En un riñón sano, la claudina-16 y la claudina-19 se localizan en las uniones entre las células de un segmento específico de los túbulos de filtrado, donde regulan con precisión cuánto magnesio y calcio se recupera de la orina en formación. Los cambios dañinos en estas proteínas debilitan esa barrera, de modo que el magnesio y el calcio se pierden en la orina en lugar de volver al torrente sanguíneo. Con el tiempo, el exceso de calcio en el tejido renal favorece la formación de cálculos y la fibrosis, mientras el cuerpo lucha por mantener el equilibrio mineral.
Cinco niñas, muchas caras de la misma enfermedad
El equipo de investigación estudió a cinco niñas de cuatro familias Han chinas no emparentadas que presentaban depósitos de calcio o cálculos en los riñones, a menudo detectados durante pruebas por otros problemas como infecciones o pubertad precoz. Todas tenían magnesio plasmático bajo y la mayoría mostraba signos tempranos de enfermedad renal crónica. Las pruebas genéticas detalladas revelaron que cuatro niñas portaban distintos pares de variantes dañinas en CLDN16, mientras que una niña tenía una combinación de una alteración previamente conocida y un cambio dañino recién descubierto en CLDN19. Algunas de estas variantes provocan que la proteína claudina quede truncada; otras modifican sutilmente su estructura de modo que deja de funcionar correctamente. Sin embargo, incluso dentro de la misma familia, los síntomas variaron en severidad, lo que subraya lo impredecible que puede ser la enfermedad renal hereditaria.
Pistas inesperadas más allá de los riñones
No todos los hallazgos se limitaron al aparato urinario. Una niña con cambios en CLDN16 desarrolló un quiste ovárico hemorrágico y signos de pubertad precoz, una combinación no vinculada previamente a este trastorno renal. Dado que las claudinas también contribuyen a sellar superficies en otros órganos, incluidas las gónadas y los ojos, los autores sugieren que la claudina-16 o la claudina-19 defectuosas podrían alterar barreras fuera del riñón de formas que aún no se comprenden completamente. Otra niña con mutaciones en CLDN19 presentó anomalías oculares detectadas más de un año antes de reconocer su enfermedad renal, lo que recuerda informes anteriores que muestran que los problemas de visión pueden ser un signo de aviso temprano de esta forma de FHHNC. En conjunto, estas historias demuestran que el mismo defecto molecular básico puede manifestarse de maneras diversas en distintos pacientes.
Por qué importa el cribado genético precoz
En la actualidad no existe una cura que repare las proteínas claudina defectuosas, por lo que el tratamiento se centra en proteger los riñones: ingesta generosa de líquidos, suplementos de magnesio y citrato, y fármacos que reducen la pérdida de calcio en la orina. Incluso con estos cuidados, muchos pacientes acaban necesitando un trasplante renal. Este estudio, el mayor análisis multifamiliar de FHHNC publicado hasta ahora en China, añade una nueva variante de CLDN19 al registro médico y documenta una inusual complicación ovárica en una niña con alteraciones en CLDN16. Para las familias y los clínicos, el mensaje es claro: en niños con hipomagnesemia inexplicada, cálculos renales o daño renal temprano—especialmente cuando hay familiares afectados—las pruebas genéticas pueden proporcionar un diagnóstico firme, guiar el seguimiento de riñones y ojos y abrir la puerta a tratamientos dirigidos emergentes en el futuro.
Cita: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Palabras clave: enfermedad renal familiar, pérdida de magnesio, mutaciones de claudina, nefrocalcinosis pediátrica, tubulopatía genética