Clear Sky Science · fr
Hypomagnésémie familiale avec hypercalciurie et néphrocalcinose causée par des mutations CLDN16/CLDN19 dans quatre familles chinoises
Quand l’équilibre minéral se dérègle dans les reins
Nos reins équilibrent discrètement des minéraux comme le magnésium et le calcium à chaque minute de notre vie. Lorsque ce processus se dérègle, des enfants peuvent développer des calculs rénaux, des cicatrices et même une insuffisance rénale. Cette étude suit cinq filles issues de quatre familles chinoises qui partagent une maladie rénale héréditaire rare perturbant cet équilibre minéral. En associant une observation clinique attentive à des tests génétiques modernes, les chercheurs ont mis au jour des anomalies génétiques connues et nouvelles, aidant les médecins à mieux reconnaître et prendre en charge cette maladie peu commune.
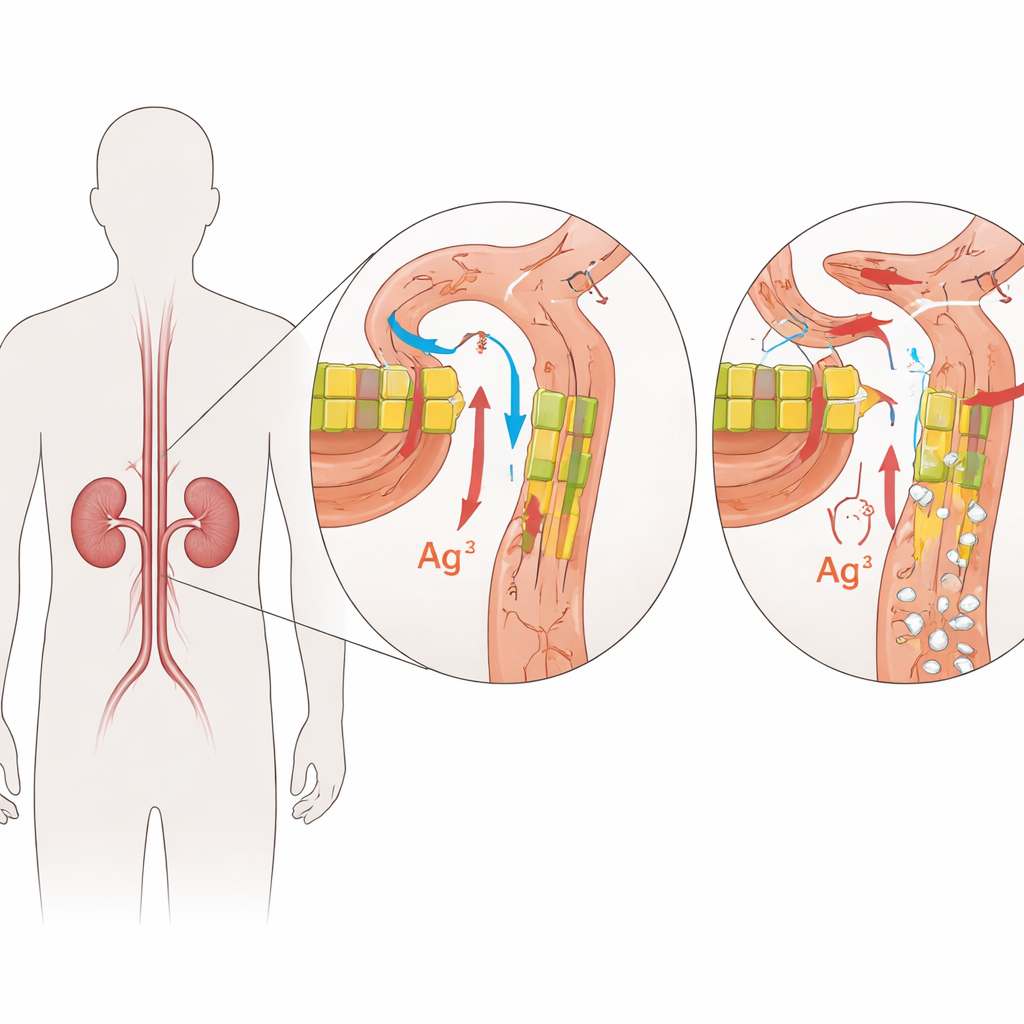
Une affection rénale familiale rare
Le trouble étudié ici s’appelle hypomagnésémie familiale avec hypercalciurie et néphrocalcinose, ou FHHNC. Les enfants atteints de FHHNC perdent trop de magnésium et de calcium dans leurs urines, ce qui conduit à une hypomagnésémie sanguine, à des dépôts riches en calcium dans les reins et à une perte progressive de la fonction rénale. Beaucoup présentent aussi une soif et une diurèse excessives ou des infections urinaires répétées. Chez certains patients, la même affection touche également les yeux, entraînant des troubles de la vision dès le plus jeune âge. La maladie se transmet selon un mode autosomique récessif : l’enfant tombe malade seulement lorsqu’il hérite d’une copie défaillante du gène concerné de chacun des parents.
Les gardiens des filtres rénaux
La FHHNC est causée par des altérations de deux gènes étroitement apparentés, CLDN16 et CLDN19. Ces gènes codent des claudines, de petites protéines qui jouent le rôle de gardiens dans les espaces étroits entre les cellules rénales. Dans un rein sain, la claudine‑16 et la claudine‑19 se situent dans les jonctions entre cellules d’un segment précis des minuscules tubules filtrants, où elles ajustent la quantité de magnésium et de calcium récupérée depuis l’urine en formation. Les altérations délétères de ces protéines affaiblissent cette barrière, si bien que le magnésium et le calcium s’échappent dans l’urine au lieu d’être restitués dans la circulation sanguine. Au fil du temps, l’excès de calcium dans le tissu rénal favorise la formation de calculs et de cicatrices, tandis que l’organisme peine à maintenir l’équilibre des minéraux.
Cinq enfants, de multiples visages d’une même maladie
L’équipe a étudié cinq filles issues de quatre familles Han chinoises non apparentées, chez qui des dépôts calciques ou des calculs rénaux ont été détectés, souvent lors d’examens pour d’autres problèmes tels que des infections ou une puberté précoce. Toutes présentaient une hypomagnésémie sanguine, et la plupart montraient des signes précoces de maladie rénale chronique. Des tests génétiques détaillés ont révélé que quatre filles portaient différentes paires de variants délétères dans CLDN16, tandis qu’une fille portait une combinaison d’une altération déjà décrite et d’une variation nouvellement identifiée dans CLDN19. Certains de ces variants entraînent l’arrêt prématuré de la protéine claudine ; d’autres modifient subtilement sa structure de sorte qu’elle ne fonctionne plus correctement. Même au sein d’une même famille, cependant, la gravité des symptômes variait, soulignant l’imprévisibilité des maladies rénales héréditaires.
Indices inattendus au‑delà des reins
Toutes les observations ne se limitaient pas aux voies urinaires. Une fille porteuse de variants CLDN16 a développé un kyste ovarien hémorragique et des signes de puberté précoce, une combinaison qui n’avait pas été associée auparavant à ce trouble rénal. Parce que les claudines contribuent aussi à sceller les surfaces d’autres organes, y compris les ovaires et les yeux, les auteurs suggèrent que des claudines‑16 ou ‑19 défectueuses pourraient perturber des barrières au‑delà du rein d’une manière encore mal comprise. Une autre fille porteuse de mutations CLDN19 présentait des anomalies oculaires détectées plus d’un an avant la reconnaissance de sa maladie rénale, rappelant des rapports antérieurs selon lesquels des troubles visuels peuvent être un signe avant‑coureur de cette forme de FHHNC. Ensemble, ces cas montrent qu’un même défaut moléculaire de base peut se manifester de façons très diverses d’un enfant à l’autre.
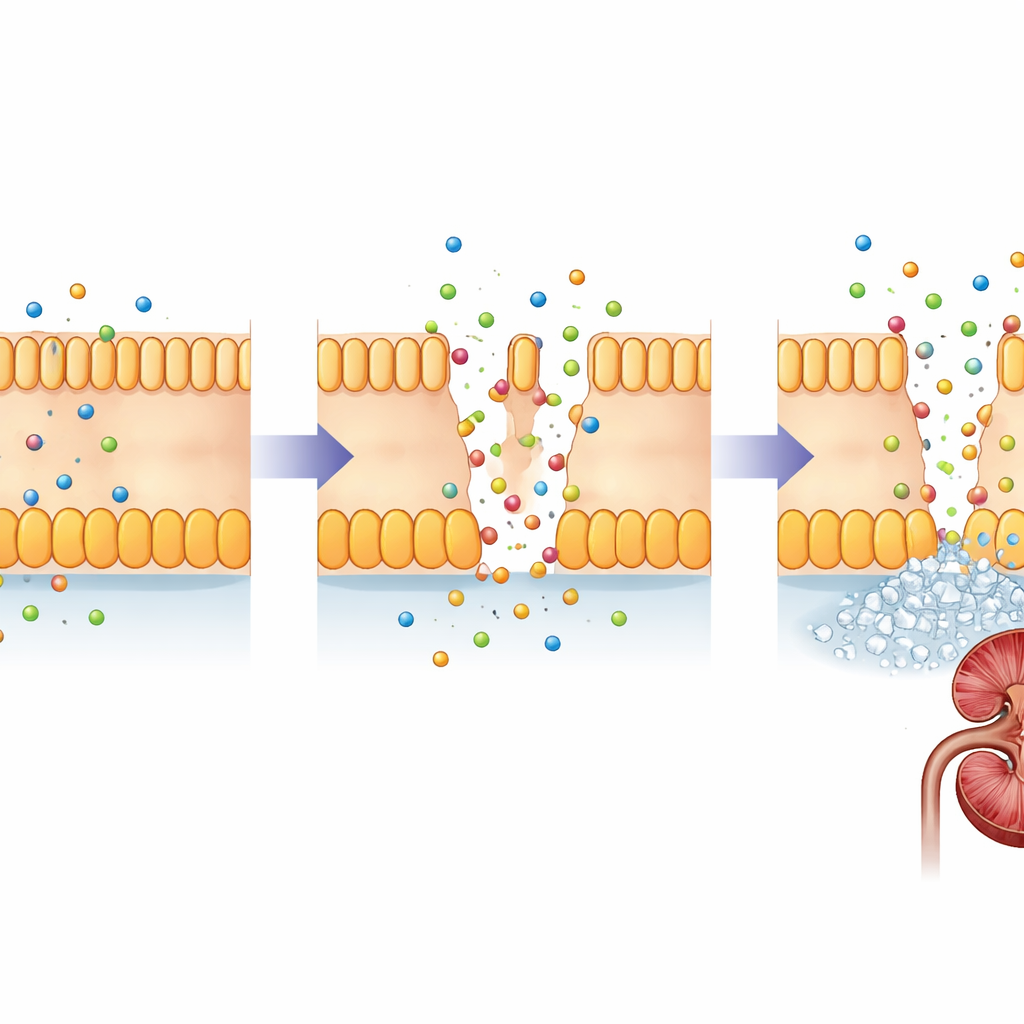
Pourquoi le dépistage génétique précoce est important
À l’heure actuelle, il n’existe pas de traitement qui répare les protéines claudines défectueuses ; la prise en charge vise donc à protéger les reins : apport hydrique généreux, suppléments de magnésium et de citrate, et médicaments réduisant la perte de calcium dans les urines. Malgré ces mesures, de nombreux patients finissent par nécessiter une transplantation rénale. Cette étude, la plus importante analyse multi‑familiale de FHHNC rapportée à ce jour en Chine, ajoute un nouveau variant CLDN19 à la littérature médicale et documente une complication ovarienne inhabituelle chez une enfant porteuse de variants CLDN16. Pour les familles et les cliniciens, le message est clair : chez les enfants présentant une hypomagnésémie inexpliquée, des calculs rénaux ou une atteinte rénale précoce — surtout lorsque des apparentés sont touchés — le diagnostic génétique peut établir un diagnostic précis, orienter la surveillance des reins et des yeux, et ouvrir la voie à des traitements ciblés émergents à l’avenir.
Citation: Wang, C., Ding, J., Yang, H. et al. Familial hypomagnesemia with hypercalciuria and nephrocalcinosis caused by CLDN16/CLDN19 mutations in four Chinese families. Sci Rep 16, 10903 (2026). https://doi.org/10.1038/s41598-026-45530-0
Mots-clés: maladie rénale familiale, perte de magnésium, mutations des claudines, néphrocalcinose pédiatrique, tubulopathie génétique