Clear Sky Science · tr
Yeni SLC16A2 mutasyonları tiroid hormonu taşımasını bozar ve Allan–Herndon–Dudley sendromlu Çinli hastalarda nörogelişimsel bozukluklara yol açar
Hormonal Bir Beyin Yolunun Tıkanması
Tiroid hormonları en çok kilo ve enerji düzenleyicileri olarak bilinir, ancak gelişmekte olan beyin için de kritik yapısal düzenleyicilerdir. Bu çalışma, Allan–Herndon–Dudley sendromu adı verilen nadir bir durumdaki bazı çocukların neden derin hareket ve öğrenme güçlükleriyle büyüdüğünü inceliyor. Araştırmacılar, belirli genetik değişikliklerin tiroid hormonunun beyine ulaşmasını nasıl engellediğini ortaya koyarak erken beyin devrelerinin gizli kimyasını aydınlatıyor ve gelecekte tasarlanabilecek tedavilere yönelik yeni yollar işaret ediyor.
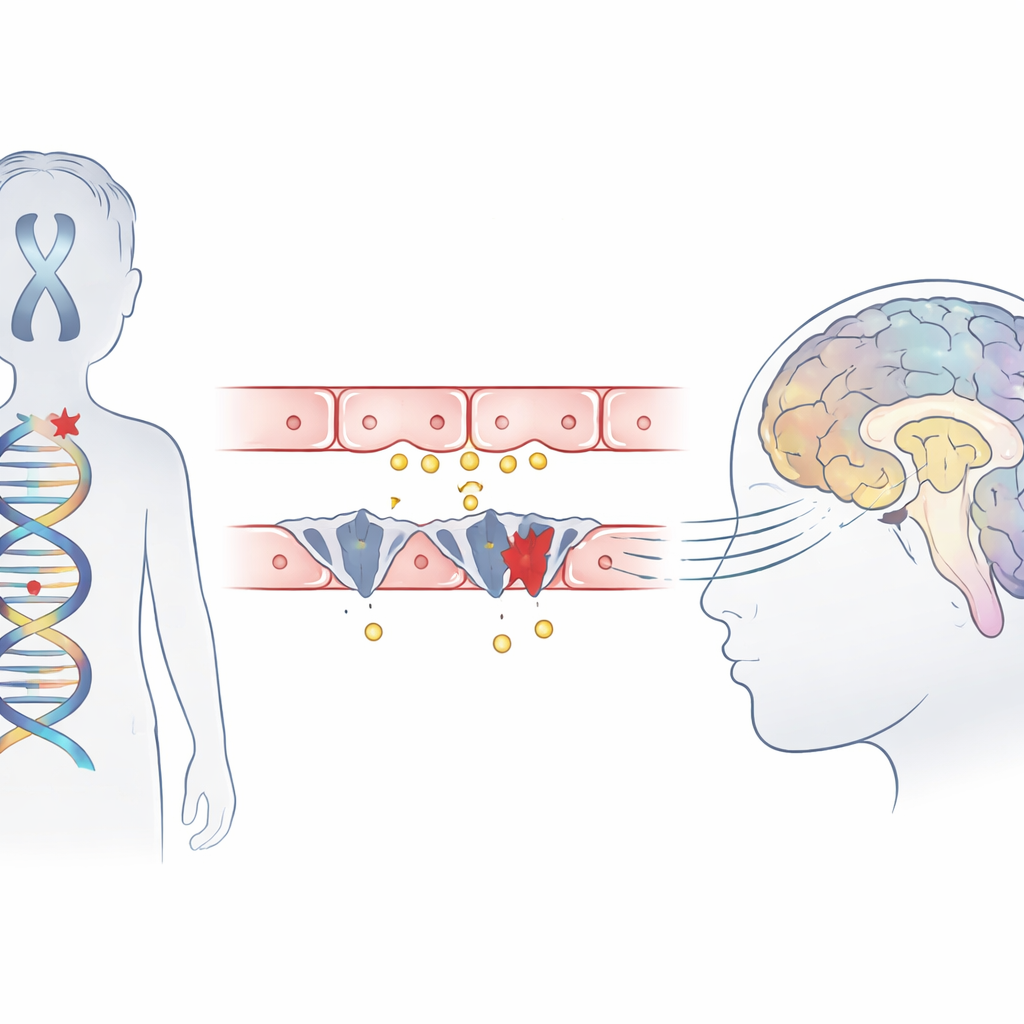
Odaklanılan Nadir Bir Çocukluk Hastalığı
Allan–Herndon–Dudley sendromu, ağırlıklı olarak erkekleri etkileyen kalıtsal bir durum olup ciddi zihinsel gerilik, bağımsız oturma veya yürüme yetisinin olmaması ve manyetik rezonans görüntülerinde görülen gecikmiş beyin bağlantıları ile seyreder. Kan testleri alışılmadık bir tiroid hormonu profili gösterse de, bezin kendisi asıl sorun değildir. Önceki çalışmalar, aktif hormonu beyin hücrelerine taşıyan MCT8 adlı taşıyıcı proteinin eksik veya işlevsiz olduğunu düşündürmüştü. Bu çalışma, alışılagelmiş sendrom semptomları gösteren ve birbirinden bağımsız üç Çinli ailenin üç çocuğunu rapor ediyor ve bu mutasyonların hormonun beyne girişini nasıl bozduğunu ayrıntılı şekilde araştırıyor.
Hormonu Engelleyen Genler
Ekzom dizilemesi kullanarak ekip çocukların DNA’sını taradı ve MCT8 taşıyıcısını kodlayan SLC16A2 geninde zararlı değişiklikler tespit etti. İki çocuk, proteini kısa kesen yeni tanımlanmış “trunkasyon” mutasyonları taşıyordu ve üçüncü çocuk ise gen parçalarının normalde birleştirildiği kritik bir eklem yerinde (splice site) mutasyon taşıyordu. Protein yapısı bilgisayar modelleri, bu değişikliklerden birinin hormon bağlanma cebini oluşturmaya yardımcı olan kritik bir helikal segmentin bir kısmını kopardığını ve taşıyıcının çalışmasını engellediğini öne sürdü. Üç varyant da geniş popülasyon veritabanlarında yoktu ve genin birçok türde güçlü biçimde korunan bölgelerinde yer alıyordu; bu da bunların gerçekten zararlı olduğuna dair kanıtı güçlendiriyor.
Kan Hücrelerindeki Sinyaller Açlıktaki Beyni Gösteriyor
Bu mutasyonların canlı hücrelerde nasıl etkili olduğunu görmek için araştırmacılar çocukların, ebeveynlerinin ve sağlıklı kontrollerin kan hücrelerinde gen aktivitesini ölçtü. Etkilenen çocuklarda SLC16A2 geninin kendisi çok daha düşük düzeyde eksprese ediliyordu; bu, hatalı proteinin yok edilmesiyle tutarlıydı. Aynı zamanda, yerel tiroid hormonu düzeylerine yanıt veren diğer genler tipik bir desen gösterdi: Hücreler hormon eksikliği algıladığında sıkça artan DIO2 ve HR yükselirken, sinir büyümesini, sinaps oluşumunu ve miyelinleşmeyi yönlendirmeye yardımcı olan Nrgn ve KIF9 keskin şekilde azalmıştı. Bu değişimler birlikte, hormon girişinin yetersiz olduğu durumlarda dokuların telafi etmeye çalıştığını ancak normal beyin gelişimini desteklemekte başarısız kaldığını gösteriyor.
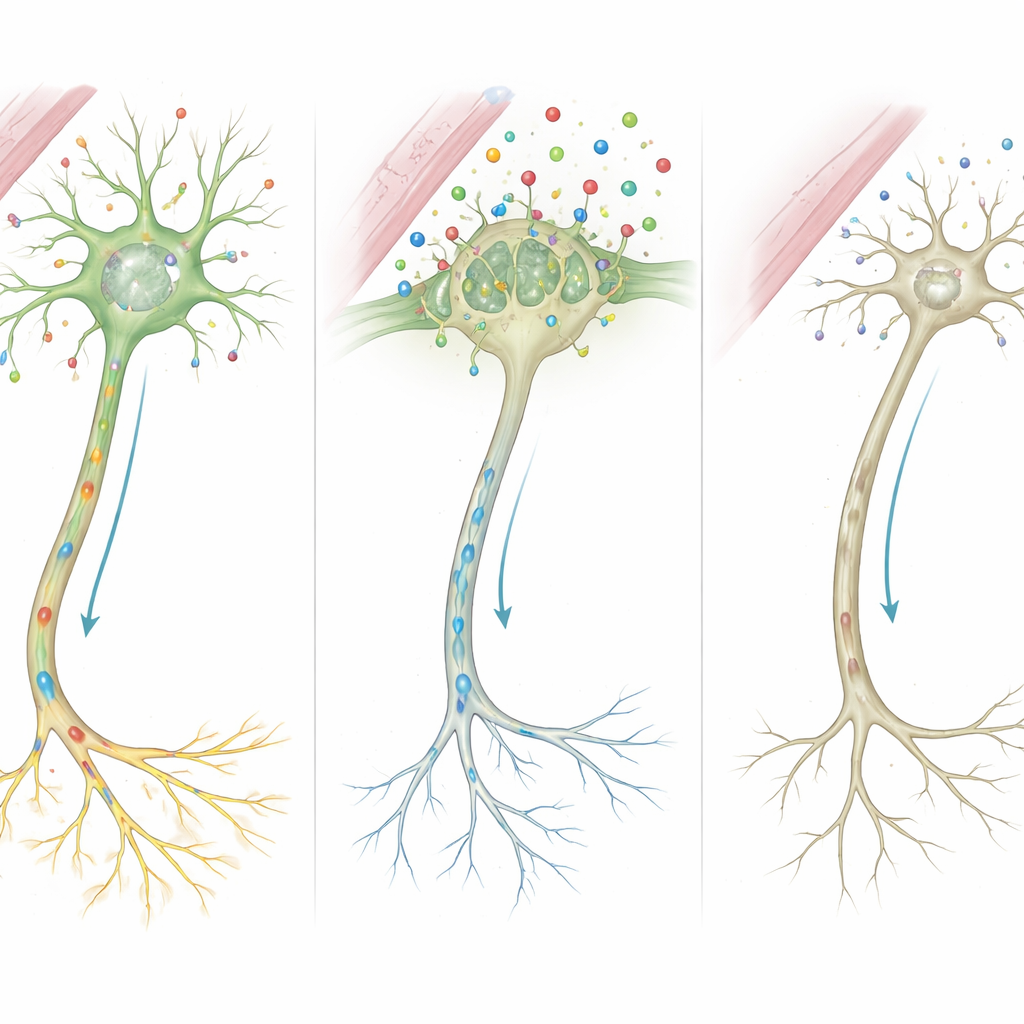
Moleküllerden Yanlış Bağlanmış Devrelere
Bu ipuçlarını birleştiren yazarlar ayrıntılı bir olay zinciri öneriyor. MCT8 eksik veya kısaltılmış olduğunda, büyümenin kritik dönemlerinde çok daha az tiroid hormonu beyin hücrelerine geçiyor. Bu sinyale mahrum kalan hücrelerin kontrol merkezi, sinir liflerinin iskeletini ve sinapsların gücünü şekillendiren genleri düzgün şekilde açıp kapatamıyor. Sinirleri yalıtan miyelin üreten destek hücreleri ve kimyasal habercileri geri toplayan hücreler de işlevlerini iyi yerine getiremiyor. Zamanla bu mikroskobik kusurlar birikerek hastalarda görülen görünür özelliklere dönüşüyor: MR’de gecikmiş miyelinleşme, kaslarda sertlik veya zayıflık, hareket bozuklukları ve şiddetli bilişsel yetersizlik.
Geleceğin Tedavileri İçin Bir Platform Oluşturmak
Üç yeni mutasyonu tanımlamanın ötesinde, çalışma açık bir bağlantıyı güçlendiriyor: SLC16A2 geni ne kadar şiddetli zarar görmüşse, engellilik o kadar derin oluyor. Ayrıca, laboratuvarda sinir hücrelerine dönüştürülebilen hasta kökenli kök hücre hatları dahil olmak üzere gelecekteki çalışmalar için bir araç seti oluşturuyor. Uzman olmayanlar için temel çıkarım şudur: beyin, tiroid hormonunun belirli bir moleküler kapı aracılığıyla zamanında taşınmasına bağımlıdır. O kapı arızalandığında gelişim kalıcı olarak aksayabilir. Bu yolun bu kadar derinlemesine anlaşılması, bir gün kan-beyin bariyerini hedefleyen, alternatif taşıyıcıları güçlendiren veya geni düzelten tedavilerin en azından bu hayati hormon yolunun bir bölümünü geri kazandırabileceği umudunu doğuruyor.
Atıf: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Anahtar kelimeler: Allan-Herndon-Dudley sendromu, tiroid hormonu taşıması, MCT8 eksikliği, nörogelişimsel bozukluklar, SLC16A2 mutasyonları