Clear Sky Science · nl
Nieuwe SLC16A2-mutaties verstoren schildklierhormoontransport en veroorzaken neuro-ontwikkelingsstoornissen bij Chinese patiënten met Allan–Herndon–Dudley-syndroom
Wanneer een hormoon-snelweg naar de hersenen hapert
Schildklierhormonen zijn vooral bekend vanwege hun rol in gewicht en energiehuishouding, maar ze zijn ook cruciale bouwmeesters van de zich ontwikkelende hersenen. Deze studie onderzoekt waarom sommige kinderen met een zeldzame aandoening, het Allan–Herndon–Dudley-syndroom, opgroeien met ernstige bewegings- en leerproblemen. Door te achterhalen hoe specifieke genetische veranderingen verhinderen dat schildklierhormoon de hersenen bereikt, werpen de onderzoekers licht op de verborgen chemie van vroege hersenontwikkeling en wijzen ze op nieuwe wegen voor mogelijke toekomstige behandelingen.
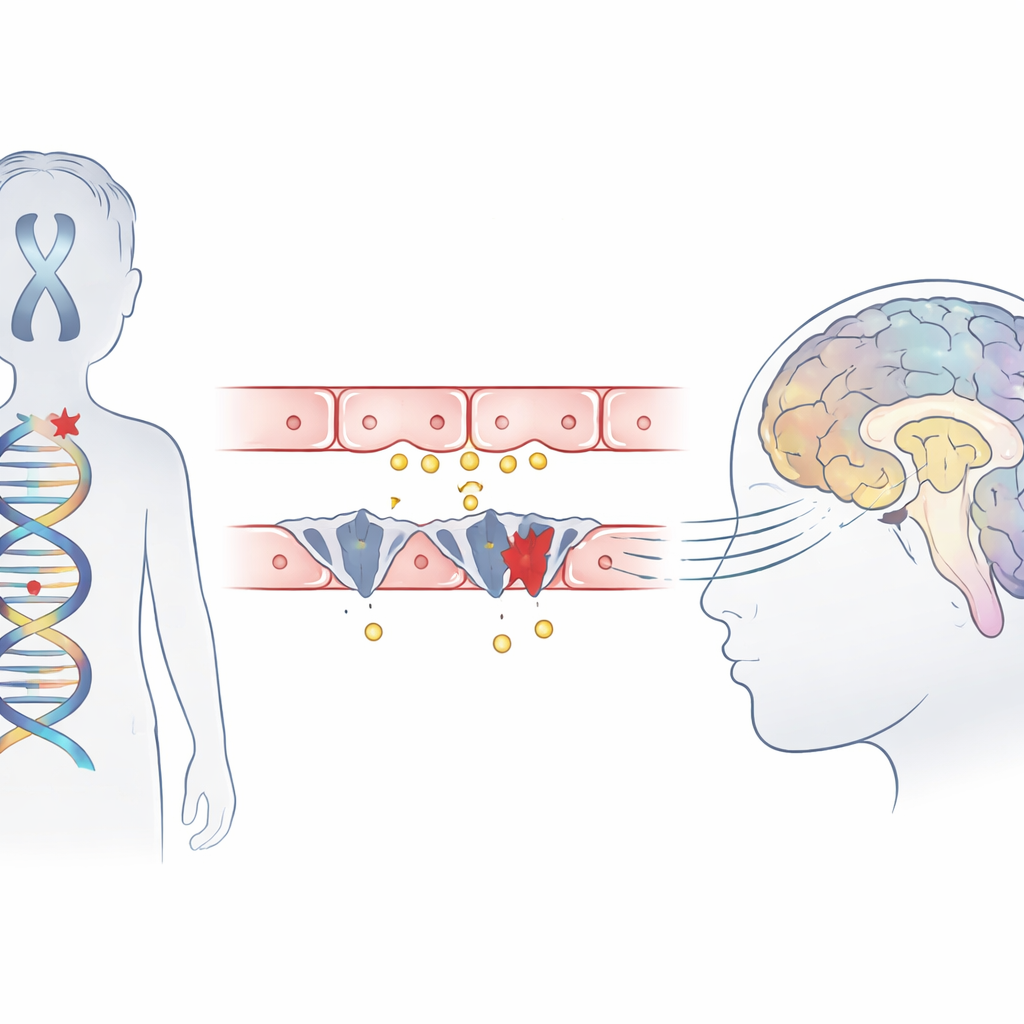
Een zeldzame kinderstoornis in de schijnwerpers
Het Allan–Herndon–Dudley-syndroom is een erfelijke aandoening die voornamelijk jongens treft en leidt tot ernstige verstandelijke beperking, het onvermogen om zelfstandig te zitten of te lopen, en vertraagde hersenontwikkeling zichtbaar op MRI-scans. Hoewel bloedtesten een afwijkend patroon van schildklierhormonen laten zien, is de schildklier zelf niet het primaire probleem. Eerder werk suggereerde dat een transportereiwit, MCT8 genoemd, dat normaal het actieve hormoon in hersencellen brengt, ontbreekt of defect is. Deze studie beschrijft drie jongens uit niet-verwante Chinese families die allemaal klassieke symptomen van het syndroom vertoonden en onderzoekt precies hoe hun mutaties het binnendringen van het hormoon in de hersenen blokkeren.
Genen die de hormoonpoort afsluiten
Middels exoomsequencing doorzocht het team het DNA van de kinderen en identificeerde schadelijke veranderingen in één gen, SLC16A2, dat codeert voor de MCT8-transporter. Twee jongens hadden nieuw ontdekte "truncerende" mutaties die het eiwit vroegtijdig afbreken, en de derde droeg een mutatie op een belangrijk splicepunt waar gensegmenten normaal worden samengevoegd. Computermodellen van de eiwitstructuur suggereerden dat een van deze veranderingen een deel van een cruciale helix wegsnijdt die bijdraagt aan het hormoon-bindingsvak, waardoor de transporter niet meer functioneert. Alle drie varianten ontbraken in grote populatiedatabases en lagen in delen van het gen die sterk geconserveerd zijn bij veel soorten, wat het bewijs versterkt dat ze echt schadelijk zijn.
Signalen in bloedcellen tonen een verhongerende hersenen
Om te zien hoe deze mutaties zich uiten in levende cellen, maten de onderzoekers genactiviteit in bloedcellen van de kinderen, hun ouders en gezonde controles. Het SLC16A2-gen zelf werd veel minder tot expressie gebracht bij de aangedane jongens, wat consistent is met afbraak van het defecte eiwit. Tegelijkertijd vertoonden andere genen die reageren op lokale schildklierhormoonniveaus een kenmerkend patroon: DIO2 en HR, die vaak omhooggaan wanneer cellen te weinig hormoon waarnemen, waren verhoogd, terwijl Nrgn en KIF9, die helpen bij geleiding van zenuwgroei, synapsvorming en myelinisatie, sterk waren verlaagd. Samen schetsen deze verschuivingen het beeld van weefsels die proberen te compenseren voor slecht hormoontoetred, maar er niet in slagen normale hersenontwikkeling te ondersteunen.
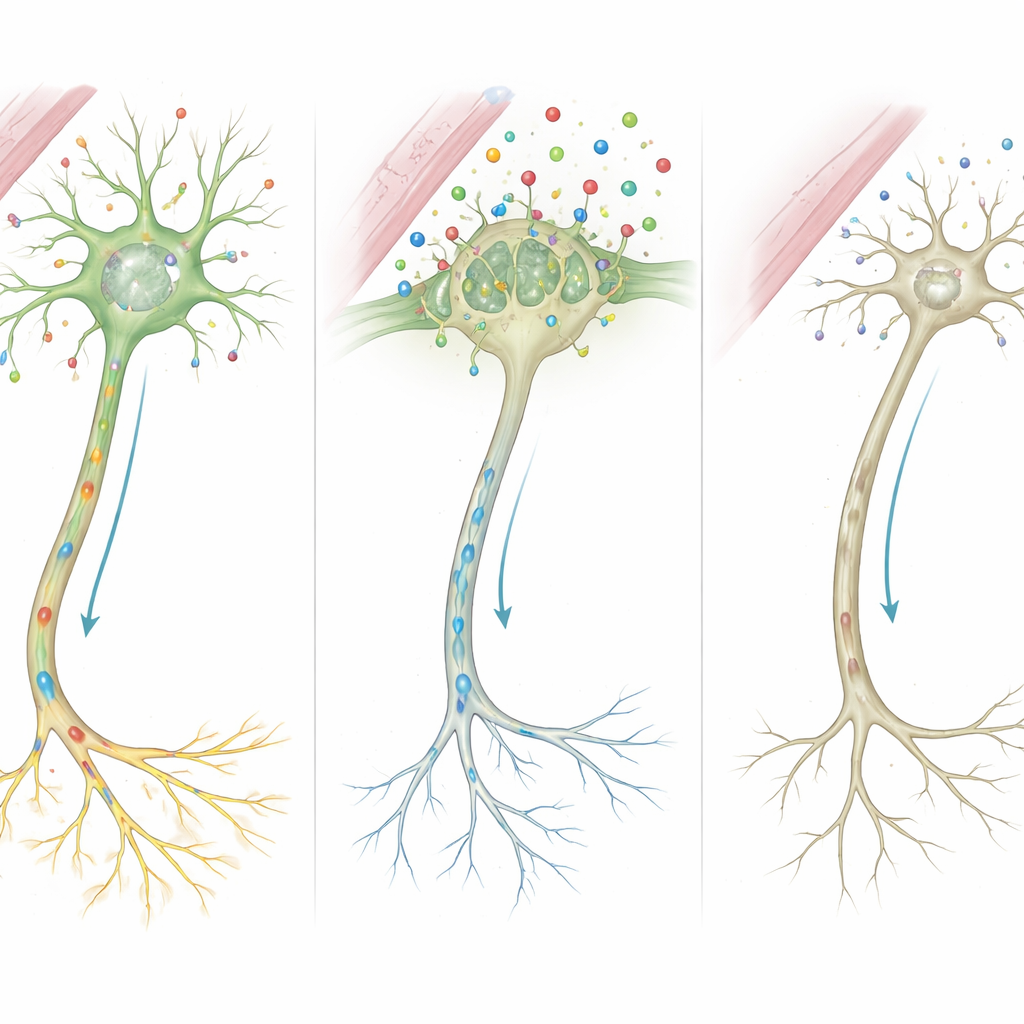
Van moleculen naar verkeerd bedrade netwerken
Als men deze aanwijzingen samenvoegt, stellen de auteurs een gedetailleerde gebeurtenissenketen voor. Omdat MCT8 ontbreekt of afgebroken is, komt veel minder schildklierhormoon tijdens cruciale groeiperioden de hersencellen binnen. Zonder dit signaal kan het cellulaire regelcentrum genen die het geraamte van zenuwvezels en de sterkte van synapsen vormen, niet adequaat inschakelen. Steuncellen die zenuwen isoleren met myeline, en cellen die chemische boodschappers recyclen, functioneren ook slecht. In de loop van de tijd lopen deze microscopische defecten op en leiden ze tot de zichtbare kenmerken bij patiënten: vertraagde myelinisatie op MRI, stijve of zwakke spieren, bewegingsstoornissen en ernstige cognitieve beperkingen.
Een platform bouwen voor toekomstige therapieën
Naast de beschrijving van drie nieuwe mutaties, versterkt de studie een duidelijke relatie: hoe ernstiger het SLC16A2-gen beschadigd is, hoe dieper de beperking. De studie levert ook een gereedschapskist voor vervolgonderzoek, waaronder patiënt-afgeleide stamcellijnen die in het laboratorium tot zenuwcellen kunnen worden gedifferentieerd. Voor niet-specialisten is de belangrijkste conclusie dat de hersenen afhankelijk zijn van tijdige levering van schildklierhormoon via een specifiek moleculair poortje. Als die poort faalt, kan de ontwikkeling blijvend worden verstoord. Door dit pad zo grondig te begrijpen ontstaat de hoop dat toekomstige therapieën — mogelijk gericht op de bloed-hersenbarrière, het stimuleren van alternatieve transporters of het corrigeren van het gen zelf — ten minste een deel van deze vitale hormoonsnelweg kunnen herstellen.
Bronvermelding: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Trefwoorden: Allan-Herndon-Dudley-syndroom, transport van schildklierhormoon, MCT8-tekort, neuro-ontwikkelingsstoornissen, SLC16A2-mutaties