Clear Sky Science · fr
Nouvelles mutations du gène SLC16A2 altèrent le transport des hormones thyroïdiennes et entraînent des troubles du neurodéveloppement chez des patients chinois atteints du syndrome d’Allan–Herndon–Dudley
Lorsque l’autoroute hormonale vers le cerveau se brise
Les hormones thyroïdiennes sont surtout connues pour réguler le poids et l’énergie, mais elles jouent aussi un rôle essentiel dans la construction du cerveau en développement. Cette étude examine pourquoi certains enfants atteints d’une maladie rare appelée syndrome d’Allan–Herndon–Dudley grandissent avec de profondes difficultés motrices et cognitives. En révélant comment des altérations génétiques spécifiques empêchent l’hormone thyroïdienne d’atteindre le cerveau, les chercheurs éclairent la chimie cachée du câblage cérébral précoce et indiquent des pistes pour la conception de futurs traitements.
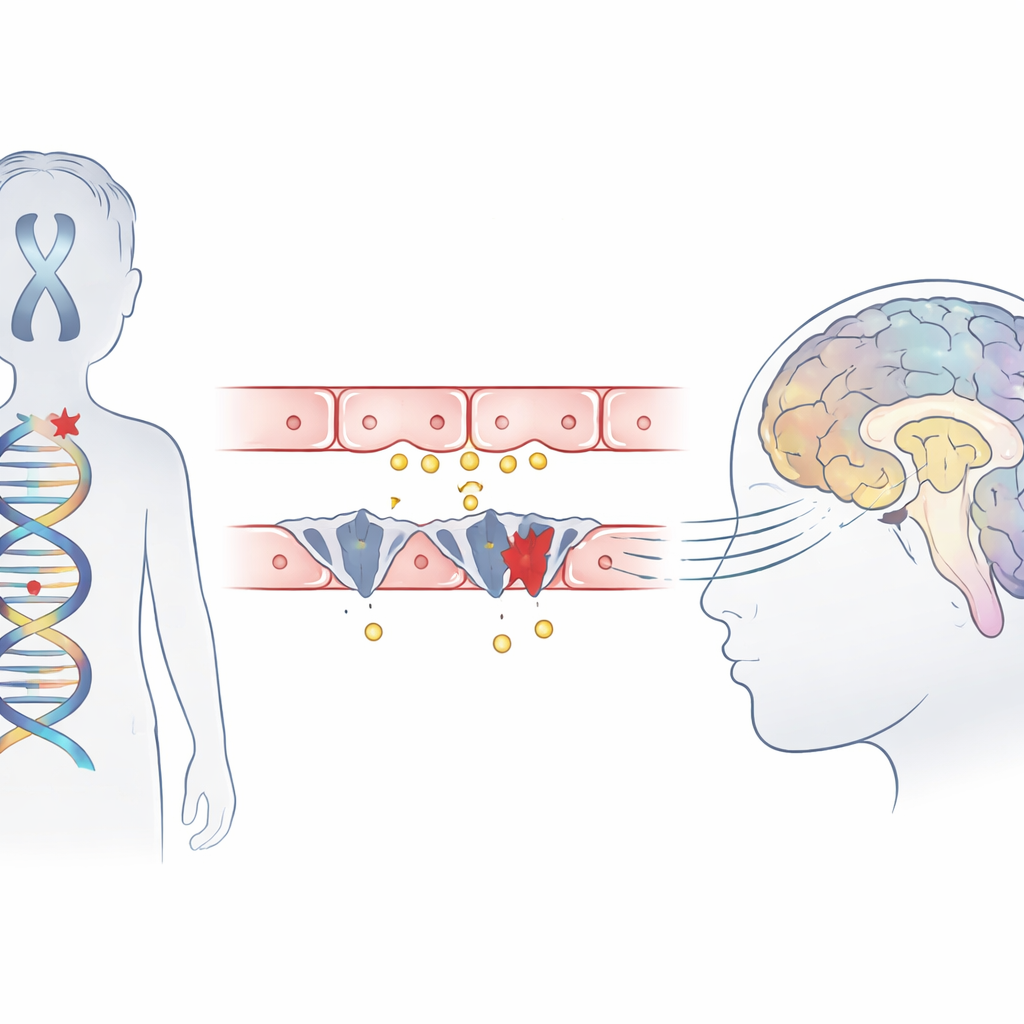
Un trouble infantile rare sur la sellette
Le syndrome d’Allan–Herndon–Dudley est une affection héréditaire qui touche principalement les garçons et conduit à une déficience intellectuelle sévère, à l’incapacité de s’asseoir ou de marcher de façon autonome, et à un retard de myélinisation visible à l’IRM. Bien que les analyses sanguines montrent un profil hormonal thyroïdien inhabituel, la glande elle-même n’est pas le problème principal. Des travaux antérieurs ont plutôt suggéré qu’une protéine transporteur, nommée MCT8, qui transporte normalement l’hormone active dans les cellules cérébrales, est absente ou défectueuse. Cette étude décrit trois garçons issus de familles chinoises non apparentées présentant les symptômes classiques du syndrome et explore précisément comment leurs mutations perturbent l’entrée de l’hormone dans le cerveau.
Des gènes qui verrouillent la porte hormonale
À l’aide du séquençage de l’exome, l’équipe a analysé l’ADN des enfants et identifié des altérations délétères dans un seul gène, SLC16A2, qui code pour le transporteur MCT8. Deux des garçons portaient des mutations « tronquantes » nouvellement découvertes qui écourtent la protéine, et le troisième présentait une mutation affectant un site d’épissage clé où les segments du gène sont normalement reliés. Les modèles informatiques de structure protéique suggèrent que l’une de ces modifications supprime une partie d’un segment hélicoïdal critique qui contribue à former la poche de liaison de l’hormone, empêchant ainsi le transporteur de fonctionner. Les trois variantes étaient absentes des grandes bases de données de population et se situaient dans des régions du gène fortement conservées chez de nombreuses espèces, renforçant l’argument en faveur de leur caractère pathogène.
Des signaux dans les cellules sanguines révèlent un cerveau en manque
Pour voir comment ces mutations se manifestent dans des cellules vivantes, les chercheurs ont mesuré l’activité génique dans les cellules sanguines des enfants, de leurs parents et de témoins sains. Le gène SLC16A2 lui‑même était exprimé à des niveaux bien plus faibles chez les garçons affectés, cohérent avec une protéine défectueuse dégradée. Parallèlement, d’autres gènes qui répondent aux niveaux locaux d’hormone thyroïdienne montraient un profil caractéristique : DIO2 et HR, qui augmentent souvent lorsque les cellules perçoivent un déficit hormonal, étaient élevés, tandis que Nrgn et KIF9, qui contribuent à guider la croissance des neurones, la formation des synapses et la myélinisation, étaient fortement réduits. Ensemble, ces changements dessinent le tableau de tissus tentant de compenser une mauvaise entrée hormonale, sans parvenir à soutenir un développement cérébral normal.
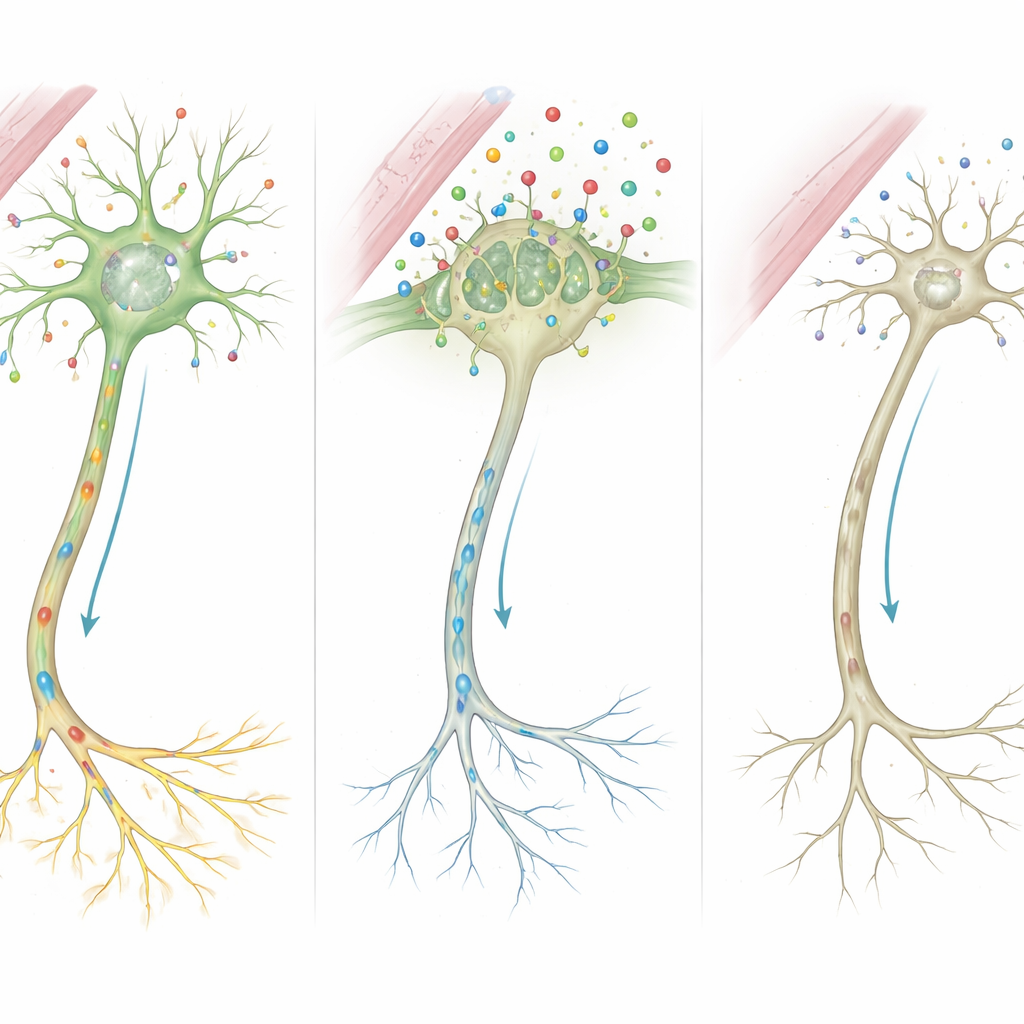
Des molécules aux circuits mal câblés
En rassemblant ces indices, les auteurs proposent une chaîne d’événements détaillée. Parce que MCT8 est absent ou tronqué, beaucoup moins d’hormone thyroïdienne traverse dans les cellules cérébrales durant des périodes critiques de croissance. Privées de ce signal, les cellules ne peuvent pas activer correctement les gènes qui façonnent l’architecture des fibres nerveuses et la force des synapses. Les cellules de soutien qui enveloppent les nerfs d’une gaine isolante de myéline, ainsi que celles qui recyclent les messagers chimiques, fonctionnent aussi mal. Avec le temps, ces défauts microscopiques s’accumulent et se traduisent par les signes observables chez les patients : retard de myélinisation à l’IRM, muscles raides ou faibles, troubles du mouvement et déficience cognitive sévère.
Poser les bases de thérapies futures
Au‑delà de la description de trois nouvelles mutations, l’étude renforce un lien clair : plus le gène SLC16A2 est sévèrement atteint, plus le handicap est profond. Elle établit également une boîte à outils pour les travaux futurs, incluant des lignées de cellules souches dérivées des patients pouvant être différenciées en neurones en laboratoire. Pour le non‑spécialiste, l’idée clé est que le cerveau dépend d’un apport opportun d’hormones thyroïdiennes via une porte moléculaire spécifique. Lorsque cette porte tombe en panne, le développement peut être irrémédiablement compromis. Comprendre cette voie en profondeur offre l’espoir que des thérapies—par exemple ciblant la barrière hémato‑encéphalique, renforçant des transporteurs alternatifs ou corrigeant le gène lui‑même—pourraient un jour rétablir au moins une partie de cette voie hormonale vitale.
Citation: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Mots-clés: Syndrome d’Allan–Herndon–Dudley, transport des hormones thyroïdiennes, déficit en MCT8, troubles du neurodéveloppement, mutations de SLC16A2