Clear Sky Science · es
Nuevas mutaciones en SLC16A2 afectan el transporte de hormonas tiroideas y provocan déficits neurodesarrollativos en pacientes chinos con el síndrome de Allan–Herndon–Dudley
Cuando una autopista hormonal hacia el cerebro se rompe
Las hormonas tiroideas son más conocidas por regular el peso y la energía, pero también son arquitectas críticas del cerebro en desarrollo. Este estudio examina por qué algunos niños con una enfermedad rara llamada síndrome de Allan–Herndon–Dudley crecen con graves dificultades de movimiento y aprendizaje. Al descubrir cómo cambios genéticos específicos bloquean el acceso de las hormonas tiroideas al cerebro, los investigadores arrojan luz sobre la química oculta del cableado cerebral temprano y señalan nuevas vías para diseñar tratamientos futuros.
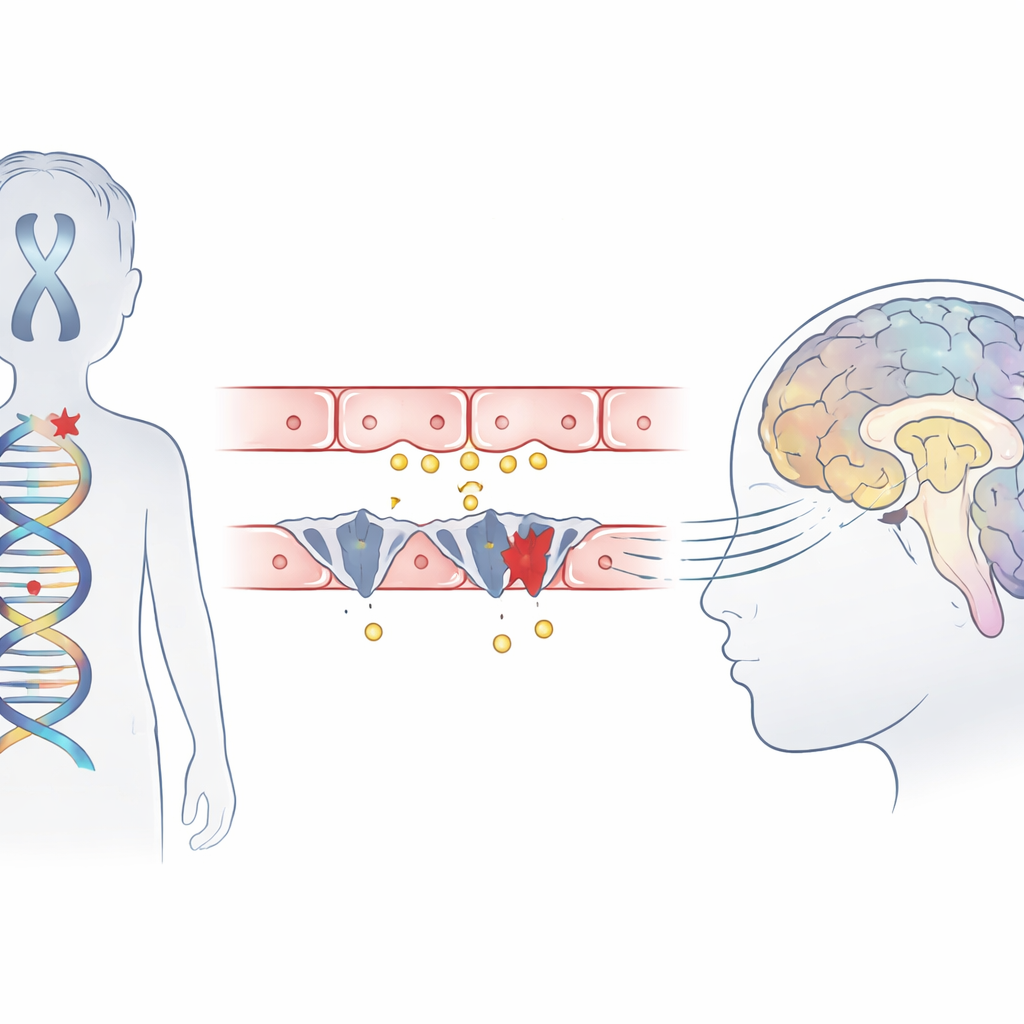
Un trastorno infantil raro en el punto de mira
El síndrome de Allan–Herndon–Dudley es una afección hereditaria que afecta principalmente a varones y provoca discapacidad intelectual grave, incapacidad para sentarse o caminar de forma independiente y un retraso en el cableado cerebral visible en las resonancias magnéticas. Aunque los análisis de sangre muestran un patrón inusual de hormonas tiroideas, la glándula en sí no es el problema principal. Trabajos previos indicaron que una proteína transportadora llamada MCT8, que normalmente transporta la hormona activa a las células cerebrales, está ausente o es defectuosa. Este estudio describe a tres niños de familias chinas no emparentadas que mostraron los síntomas clásicos del síndrome y explora cómo sus mutaciones interfieren exactamente con la entrada de la hormona al cerebro.
Genes que bloquean la puerta hormonal
Con secuenciación del exoma, el equipo buscó en el ADN de los niños y halló cambios dañinos en un único gen, SLC16A2, que codifica al transportador MCT8. Dos niños presentaban mutaciones "truncantes" recién identificadas que acortan la proteína, y el tercero tenía una mutación en un sitio de empalme clave donde normalmente se unen los segmentos del gen. Modelos computacionales de la estructura proteica sugirieron que uno de estos cambios elimina parte de un segmento helicoidal crítico que contribuye a formar el bolsillo de unión de la hormona, impidiendo que el transportador funcione. Las tres variantes estaban ausentes en grandes bases de datos poblacionales y se situaban en regiones del gen fuertemente conservadas entre muchas especies, lo que refuerza la hipótesis de que son realmente perjudiciales.
Señales en las células sanguíneas revelan un cerebro hambriento
Para ver cómo se manifiestan estas mutaciones en células vivas, los investigadores midieron la actividad génica en células sanguíneas de los niños, sus padres y controles sanos. El propio gen SLC16A2 presentaba una expresión mucho más baja en los varones afectados, coherente con la degradación de la proteína defectuosa. Al mismo tiempo, otros genes que responden a los niveles locales de hormonas tiroideas mostraron un patrón revelador: DIO2 y HR, que suelen aumentar cuando las células perciben escasez hormonal, estaban elevados, mientras que Nrgn y KIF9, que ayudan a guiar el crecimiento neuronal, la formación de sinapsis y la mielinización, estaban marcadamente reducidos. En conjunto, estos cambios describen tejidos que intentan compensar la mala entrada hormonal, pero fracasan en apoyar un desarrollo cerebral normal.
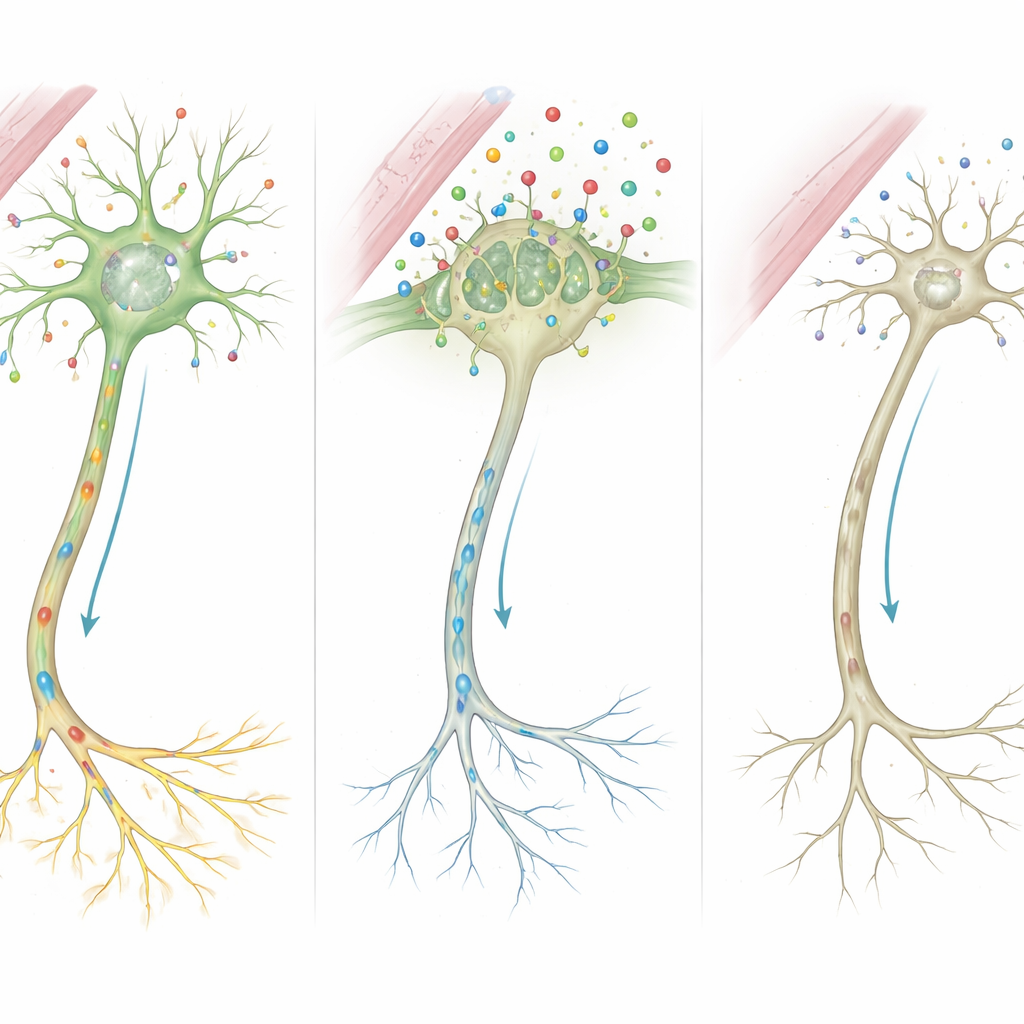
De las moléculas a los circuitos mal cableados
Al juntar estas pistas, los autores proponen una cadena de eventos detallada. Debido a que MCT8 está ausente o truncado, entra mucha menos hormona tiroidea en las células cerebrales durante periodos críticos de crecimiento. Privadas de esa señal, las células no activan correctamente genes que moldean el esqueleto de las fibras nerviosas y la fortaleza de las sinapsis. Las células de soporte que recubren los nervios con mielina y las que reciclan mensajeros químicos también funcionan mal. Con el tiempo, estos defectos microscópicos se acumulan y dan lugar a las características visibles en los pacientes: mielinización retrasada en la resonancia, músculos rígidos o débiles, trastornos del movimiento y deterioro cognitivo severo.
Construyendo una plataforma para terapias futuras
Más allá de describir tres nuevas mutaciones, el estudio refuerza un vínculo claro: cuanto más gravemente está dañado el gen SLC16A2, más profunda es la discapacidad. También establece una caja de herramientas para trabajos futuros, incluidas líneas celulares madre derivadas de pacientes que pueden convertirse en neuronas en el laboratorio. Para los no especialistas, la conclusión clave es que el cerebro depende de la entrega oportuna de la hormona tiroidea a través de una puerta molecular específica. Cuando esa puerta falla, el desarrollo puede desviarse de forma permanente. Entender esta vía en tal detalle ofrece la esperanza de que algún día terapias —quizá dirigidas a la barrera hematoencefálica, potenciando transportadores alternativos o corrigiendo el gen mismo— puedan restaurar al menos parte de esta vital autopista hormonal.
Cita: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Palabras clave: Síndrome de Allan–Herndon–Dudley, transporte de hormonas tiroideas, deficiencia de MCT8, trastornos del neurodesarrollo, mutaciones en SLC16A2