Clear Sky Science · pt
Novas mutações em SLC16A2 prejudicam o transporte de hormônio tireoidiano e provocam déficits do neurodesenvolvimento em pacientes chineses com síndrome de Allan–Herndon–Dudley
Quando uma via hormonal para o cérebro se rompe
Os hormônios tireoidianos são mais conhecidos por controlar peso e energia, mas também são arquitetos essenciais do cérebro em desenvolvimento. Este estudo examina por que algumas crianças com uma condição rara chamada síndrome de Allan–Herndon–Dudley crescem com dificuldades profundas de movimento e aprendizagem. Ao descobrir como alterações genéticas específicas bloqueiam a chegada do hormônio tireoidiano ao cérebro, os pesquisadores lançam luz sobre a química oculta da formação cerebral precoce e apontam para novas abordagens que tratamentos futuros podem adotar.
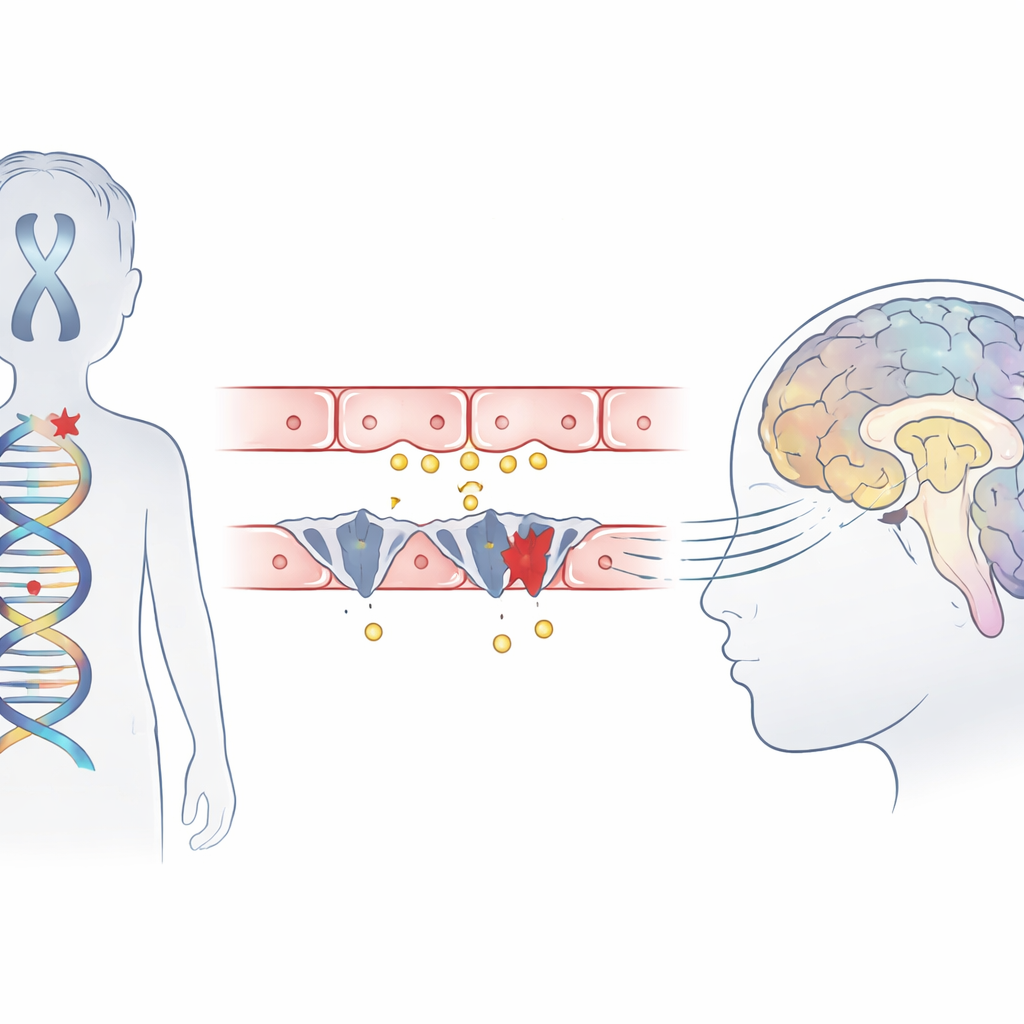
Um transtorno infantil raro em foco
A síndrome de Allan–Herndon–Dudley é uma condição hereditária que afeta principalmente meninos e leva a deficiência intelectual grave, incapacidade de sentar ou andar de forma independente e atraso na formação das conexões cerebrais observadas em ressonância magnética. Embora exames de sangue mostrem um padrão incomum de hormônio tireoidiano, a própria glândula não é o problema principal. Em vez disso, trabalhos anteriores sugeriram que uma proteína transportadora, chamada MCT8, que normalmente leva o hormônio ativo para dentro das células do cérebro, está ausente ou defeituosa. Este estudo descreve três meninos de famílias chinesas não relacionadas que apresentaram sintomas clássicos da síndrome e explora exatamente como suas mutações interferem na entrada do hormônio no cérebro.
Genes que bloqueiam o portão hormonal
Usando sequenciamento de exoma, a equipe vasculhou o DNA das crianças e identificou alterações deletérias em um único gene, SLC16A2, que codifica o transportador MCT8. Dois meninos carregavam mutações truncantes recém-descobertas que encurtam a proteína, e o terceiro tinha uma mutação em um sítio de splicing-chave onde segmentos do gene normalmente são unidos. Modelos computacionais da estrutura proteica sugeriram que uma dessas alterações elimina parte de um segmento helicoidal crítico que ajuda a formar o bolso de ligação ao hormônio, impedindo o funcionamento do transportador. Todas as três variantes estavam ausentes em grandes bancos de dados populacionais e situavam-se em regiões do gene altamente conservadas entre muitas espécies, reforçando a hipótese de que são realmente prejudiciais.
Sinais em células sanguíneas revelam um cérebro faminto
Para avaliar como essas mutações se manifestam em células vivas, os pesquisadores mediram a atividade gênica em células sanguíneas das crianças, de seus pais e de controles saudáveis. O próprio gene SLC16A2 foi expresso em níveis muito mais baixos nos meninos afetados, consistente com a degradação da proteína defeituosa. Ao mesmo tempo, outros genes que respondem aos níveis locais de hormônio tireoidiano exibiram um padrão característico: DIO2 e HR, que frequentemente aumentam quando as células detectam hormônio em falta, estavam elevados, enquanto Nrgn e KIF9, que ajudam a guiar o crescimento neuronal, a formação de sinapses e a mielinização, estavam fortemente reduzidos. Em conjunto, essas alterações desenham o retrato de tecidos tentando compensar a pobre entrada de hormônio, mas sem conseguir sustentar o desenvolvimento cerebral normal.
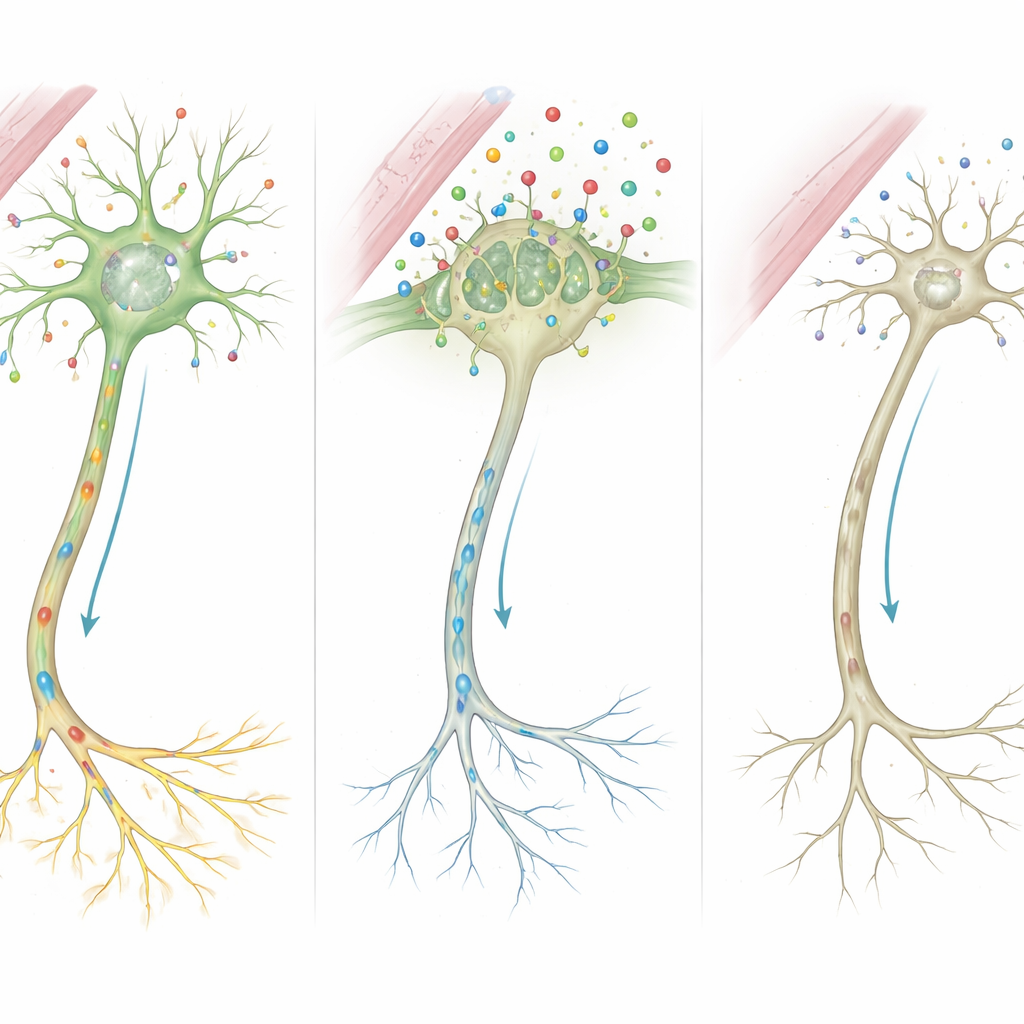
De moléculas a circuitos mal conectados
Reunindo essas pistas, os autores propõem uma cadeia de eventos detalhada. Como o MCT8 está ausente ou truncado, muito menos hormônio tireoidiano atravessa para as células cerebrais durante períodos críticos de crescimento. Privadas desse sinal, as células não conseguem ativar corretamente genes que moldam o arcabouço das fibras nervosas e a força das sinapses. Células de suporte que envolvem os nervos com mielina isolante, e aquelas que reciclam mensageiros químicos, também funcionam mal. Ao longo do tempo, esses defeitos microscópicos se acumulam nas características visíveis observadas nos pacientes: mielinização retardada na ressonância, músculos rígidos ou fracos, distúrbios do movimento e comprometimento cognitivo severo.
Construindo uma plataforma para terapias futuras
Além de descrever três novas mutações, o estudo reforça um vínculo claro: quanto mais severamente o gene SLC16A2 é danificado, mais profunda é a deficiência. Também estabelece um conjunto de ferramentas para trabalhos futuros, incluindo linhagens de células-tronco derivadas de pacientes que podem ser transformadas em neurônios no laboratório. Para não especialistas, a principal conclusão é que o cérebro depende da entrega oportuna do hormônio tireoidiano por um portão molecular específico. Quando esse portão falha, o desenvolvimento pode ser permanentemente prejudicado. Compreender essa via em tão grande detalhe oferece esperança de que um dia terapias — talvez atuando na barreira sangue–cérebro, reforçando transportadores alternativos ou corrigindo o próprio gene — possam restaurar pelo menos parte dessa via vital de hormônio.
Citação: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Palavras-chave: Síndrome de Allan–Herndon–Dudley, transporte de hormônio tireoidiano, deficiência de MCT8, transtornos do neurodesenvolvimento, mutações em SLC16A2