Clear Sky Science · it
Nuove mutazioni di SLC16A2 compromettono il trasporto di ormoni tiroidei e causano deficit dello sviluppo neurologico in pazienti cinesi con sindrome di Allan–Herndon–Dudley
Quando un’autostrada ormonale verso il cervello si interrompe
Gli ormoni tiroidei sono più noti per il loro ruolo nel controllo del peso e dell’energia, ma sono anche architetti fondamentali del cervello in sviluppo. Questo studio indaga perché alcuni bambini con una rara condizione chiamata sindrome di Allan–Herndon–Dudley crescono con gravi difficoltà motorie e di apprendimento. Scoprendo come cambiamenti genetici specifici impediscano agli ormoni tiroidei di raggiungere il cervello, i ricercatori fanno luce sulla chimica nascosta del cablaggio cerebrale precoce e indicano possibili strade per progettare terapie future.
Un raro disturbo dell’infanzia sotto i riflettori
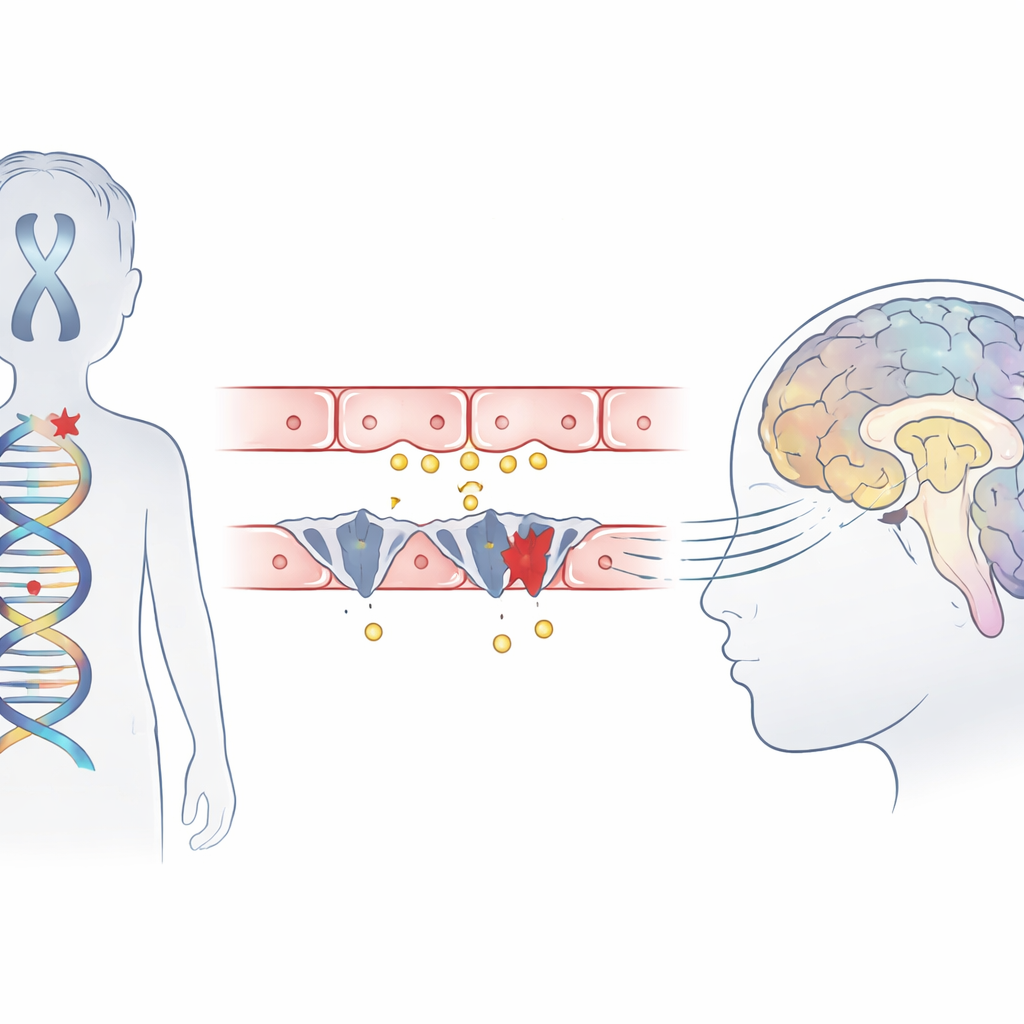
La sindrome di Allan–Herndon–Dudley è una condizione ereditaria che colpisce principalmente i ragazzi e porta a grave disabilità intellettiva, incapacità di sedersi o camminare in modo indipendente e ritardato cablaggio cerebrale visibile nelle risonanze magnetiche. Sebbene gli esami del sangue mostrino un profilo ormonale tiroideo atipico, la ghiandola non è il problema principale. Studi precedenti avevano suggerito che una proteina trasportatrice, chiamata MCT8, che normalmente trasporta l’ormone attivo nelle cellule cerebrali, sia assente o difettosa. Questo studio descrive tre ragazzi di famiglie cinesi non correlate che presentavano tutti i sintomi classici della sindrome ed esplora esattamente come le loro mutazioni interferiscano con l’ingresso dell’ormone nel cervello.
Geni che bloccano la porta ormonale
Utilizzando il sequenziamento dell’esoma, il team ha analizzato il DNA dei bambini e ha identificato variazioni dannose in un singolo gene, SLC16A2, che codifica per il trasportatore MCT8. Due ragazzi portavano nuove mutazioni “troncanti” che accorciano la proteina, mentre il terzo aveva una mutazione in un sito di splicing chiave dove i segmenti genici vengono normalmente uniti. Modelli computazionali della struttura proteica hanno suggerito che una di queste alterazioni elimina parte di un segmento elicoidale critico che contribuisce a formare la tasca di legame dell’ormone, impedendo il funzionamento del trasportatore. Tutte e tre le varianti erano assenti nei grandi database di popolazione e si trovavano in regioni del gene fortemente conservate attraverso molte specie, rafforzando l’ipotesi che siano realmente patogene.
Segnali nelle cellule del sangue rivelano un cervello in carenza
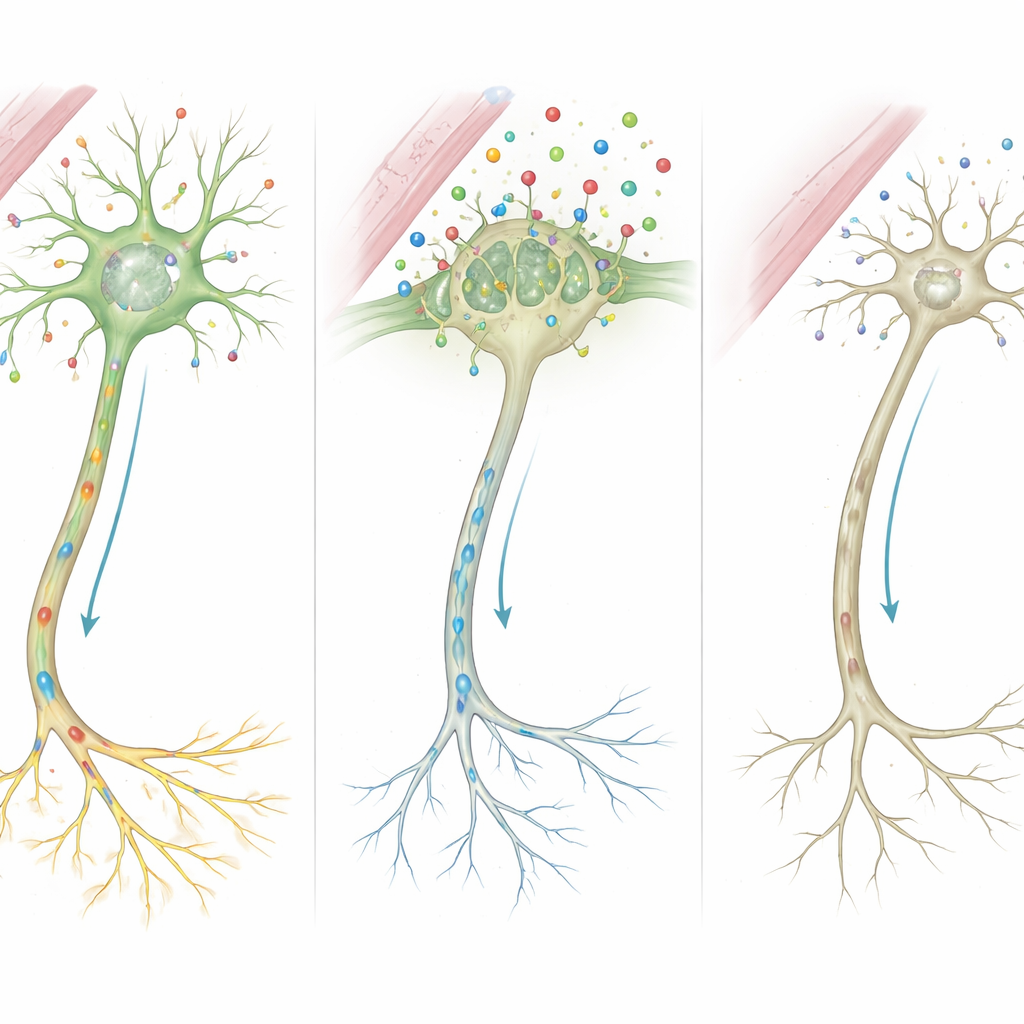
Per capire come queste mutazioni si manifestano nelle cellule viventi, i ricercatori hanno misurato l’attività genica nelle cellule del sangue dei bambini, dei loro genitori e di controlli sani. Il gene SLC16A2 stesso risultava espresso a livelli molto più bassi nei ragazzi affetti, coerentemente con la degradazione della proteina difettosa. Allo stesso tempo, altri geni che rispondono ai livelli locali di ormoni tiroidei mostravano un quadro caratteristico: DIO2 e HR, che spesso aumentano quando le cellule percepiscono carenza di ormone, erano incrementati, mentre Nrgn e KIF9, che aiutano a guidare la crescita nervosa, la formazione delle sinapsi e la mielinizzazione, risultavano fortemente ridotti. Nel complesso, questi cambiamenti dipingono il quadro di tessuti che cercano di compensare il scarso ingresso di ormone, ma non riescono a sostenere uno sviluppo cerebrale normale.
Dalle molecole ai circuiti mal cablati
Mettendo insieme questi indizi, gli autori propongono una catena di eventi dettagliata. Poiché MCT8 è assente o troncato, molta meno quantità di ormone tiroideo attraversa nelle cellule cerebrali durante periodi cruciali di crescita. Privata di questo segnale, la centrale di controllo della cellula non riesce ad attivare correttamente i geni che modellano lo scheletro delle fibre nervose e la forza delle sinapsi. Anche le cellule di supporto che avvolgono i nervi con la mielina isolante, e quelle che riciclano i messaggeri chimici, funzionano male. Nel tempo, questi difetti microscopici si accumulano fino a dare le caratteristiche visibili nei pazienti: mielinizzazione ritardata alla risonanza, muscoli rigidi o deboli, disturbi del movimento e grave compromissione cognitiva.
Costruire una piattaforma per terapie future
Oltre a descrivere tre nuove mutazioni, lo studio rafforza un legame chiaro: quanto più gravemente il gene SLC16A2 è danneggiato, tanto più profonda è la disabilità. Stabilisce inoltre una cassetta degli attrezzi per lavori futuri, incluse linee di cellule staminali derivate dai pazienti che possono essere trasformate in neuroni in laboratorio. Per i non specialisti, il messaggio chiave è che il cervello dipende dalla consegna tempestiva degli ormoni tiroidei attraverso un portale molecolare specifico. Quando quel portale fallisce, lo sviluppo può essere compromesso in modo permanente. Comprendere a fondo questa via offre speranza che un giorno terapie — magari mirate alla barriera emato-encefalica, al potenziamento di trasportatori alternativi o alla correzione del gene stesso — possano ripristinare almeno in parte questa vitale autostrada ormonale.
Citazione: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Parole chiave: Sindrome di Allan-Herndon-Dudley, trasporto di ormoni tiroidei, deficit di MCT8, disturbi dello sviluppo neurologico, mutazioni SLC16A2