Clear Sky Science · pl
Nowe mutacje SLC16A2 upośledzają transport hormonów tarczycy i powodują deficyty neuro‑rozwojowe u chińskich pacjentów z zespołem Allan–Herndon–Dudley
Gdy hormonalna autostrada do mózgu przestaje działać
Hormony tarczycy są powszechnie kojarzone z regulacją masy ciała i energii, ale pełnią też kluczową rolę jako architekci rozwijającego się mózgu. W badaniu tym analizuje się, dlaczego niektóre dzieci z rzadkim schorzeniem zwanym zespołem Allan–Herndon–Dudley dorastają z głębokimi zaburzeniami ruchu i uczenia się. Odkrywając, w jaki sposób konkretne zmiany genetyczne blokują dostęp hormonów tarczycy do mózgu, autorzy rzucają światło na ukrytą chemię wczesnego okablowania mózgu i wskazują możliwe kierunki dla przyszłych terapii.
Rzadkie dziecięce schorzenie w centrum uwagi
Zespół Allan–Herndon–Dudley to choroba dziedziczna, która głównie dotyka chłopców i prowadzi do ciężkiej niepełnosprawności intelektualnej, braku zdolności samodzielnego siedzenia lub chodzenia oraz opóźnionego okablowania mózgu widocznego w badaniach MRI. Choć badania krwi pokazują nietypowy wzorzec hormonów tarczycy, sama gruczoł nie jest głównym źródłem problemu. Wcześniejsze prace sugerowały zamiast tego defekt białka transportującego, zwanego MCT8, które normalnie dostarcza aktywny hormon do komórek mózgu. W tym badaniu opisano trzech chłopców z niepowiązanych chińskich rodzin, u których stwierdzono klasyczne objawy zespołu, i przeanalizowano, w jaki sposób ich mutacje uniemożliwiają wnikanie hormonu do mózgu.
Geny zamykające bramę dla hormonu
Wykorzystując sekwencjonowanie egzomu, zespół przeanalizował DNA dzieci i zidentyfikował uszkadzające zmiany w jednym genie, SLC16A2, który koduje transporter MCT8. Dwóch chłopców miało nowo odkryte mutacje „truncujące”, skracające białko, a trzeci nosił mutację w kluczowym miejscu splicingu, gdzie łączone są segmenty genu. Modele komputerowe struktury białka sugerowały, że jedna z tych zmian usuwa część krytycznego segmentu helikalnego, który tworzy kieszeń wiążącą hormon, uniemożliwiając prawidłowe działanie transportera. Wszystkie trzy warianty nie występowały w dużych bazach populacyjnych i leżały w częściach genu silnie zachowanych u wielu gatunków, co wzmacnia dowody na ich szkodliwość.
Sygnały w komórkach krwi ujawniają głodujący mózg
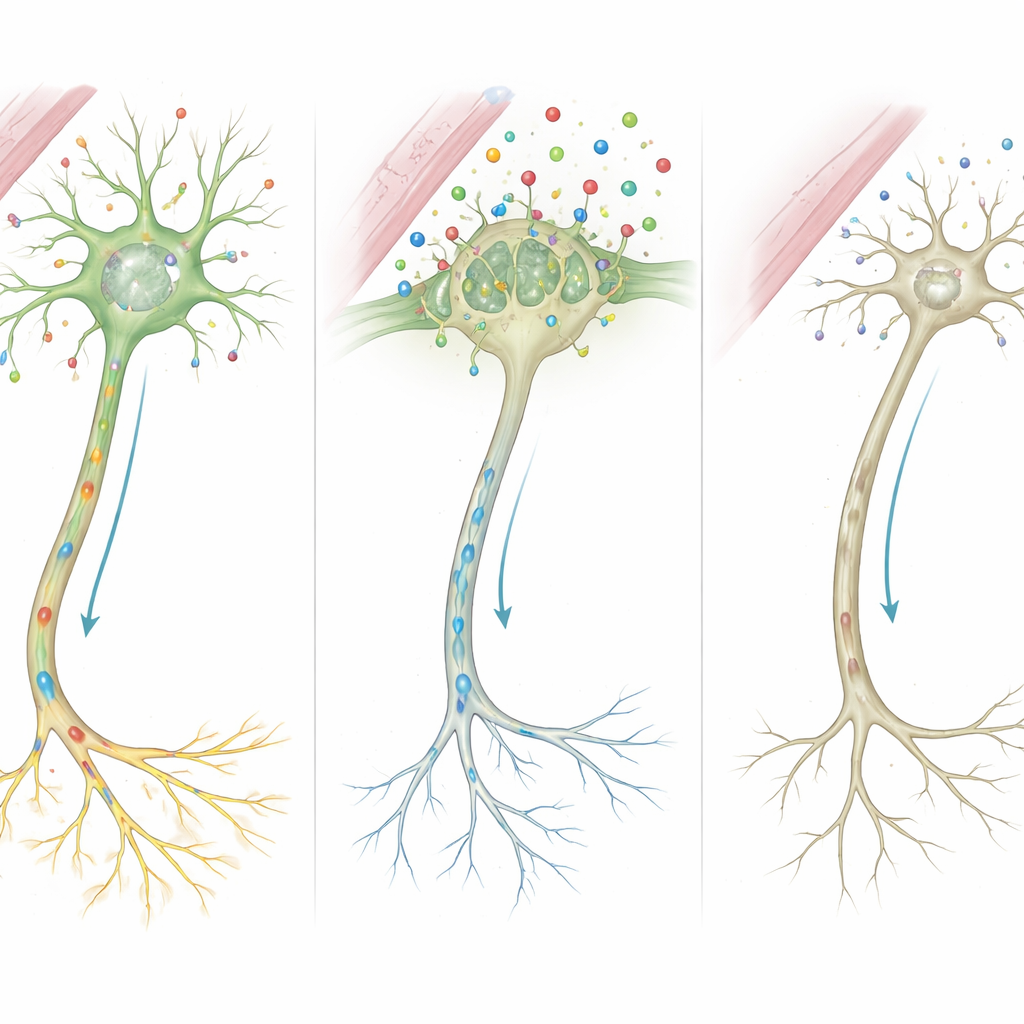
Aby zobaczyć, jak te mutacje przejawiają się w żywych komórkach, badacze zmierzyli aktywność genów w komórkach krwi dzieci, ich rodziców i zdrowych kontrolach. Sam gen SLC16A2 był znacznie niżej eksprymowany u dotkniętych chłopców, co zgadza się z hipotezą, że uszkodzone białko jest degradowane. Jednocześnie inne geny reagujące na lokalne poziomy hormonów tarczycy wykazały charakterystyczny wzorzec: DIO2 i HR, które często zwiększają ekspresję przy niedoborze hormonu, były podwyższone, podczas gdy Nrgn i KIF9, warunkujące rozwój włókien nerwowych, tworzenie synaps i mielinizację, były wyraźnie zmniejszone. Te zmiany razem tworzą obraz tkanek próbujących kompensować słaby napływ hormonu, lecz niezdolnych do podtrzymania prawidłowego rozwoju mózgu.
Od molekuł do nieprawidłowo okablowanych obwodów
Składając te wskazówki w całość, autorzy proponują szczegółowy łańcuch wydarzeń. Ponieważ MCT8 jest nieobecny lub skrócony, znacznie mniej hormonu tarczycy przedostaje się do komórek mózgu w krytycznych okresach wzrostu. Pozbawione tego sygnału, centrum kontroli komórkowej nie może prawidłowo włączać genów kształtujących szkielet włókien nerwowych i siłę synaps. Komórki wspierające, które otaczają włókna izolacją mielinową, oraz te odpowiadające za recykling przekaźników chemicznych, także działają nieprawidłowo. Z czasem te mikroskopowe defekty kumulują się i przekładają na widoczne cechy u pacjentów: opóźnioną mielinizację w MRI, sztywne lub słabe mięśnie, zaburzenia ruchu i ciężkie upośledzenie poznawcze.
Budowanie platformy dla przyszłych terapii
Poza opisaniem trzech nowych mutacji, badanie wzmacnia oczywisty wniosek: im poważniej uszkodzony jest gen SLC16A2, tym głębsza niepełnosprawność. Ustanawia także zestaw narzędzi do przyszłych prac, w tym linie komórek macierzystych pochodzących od pacjentów, które można różnicować w komórki nerwowe w laboratorium. Dla osób niebędących specjalistami kluczowy wniosek jest taki, że mózg zależy od terminowego dostarczenia hormonu tarczycy przez specyficzną molekularną bramę. Gdy ta brama zawiedzie, rozwój może zostać trwale zaburzony. Dogłębne zrozumienie tej ścieżki daje nadzieję, że kiedyś terapie — być może skierowane na barierę krew–mózg, wzmacniające alternatywne transportery lub korygujące sam gen — będą mogły przywrócić przynajmniej część tej istotnej hormonalnej autostrady.
Cytowanie: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
Słowa kluczowe: Zespół Allan–Herndon–Dudley, transport hormonów tarczycy, niedobór MCT8, zaburzenia neurorozwojowe, mutacje SLC16A2