Clear Sky Science · ar
طفرات جديدة في SLC16A2 تعيق نقل هرمون الغدة الدرقية وتسبب عجزات نمائية عصبية لدى مرضى صينيين بمتلازمة ألان-هيرندون-دادلي
عندما ينكسر طريق الهرمون إلى الدماغ
تشتهر هرمونات الغدة الدرقية بضبط الوزن والطاقة، لكنها أيضاً مهندسون أساسيون للمخ النامي. تفحص هذه الدراسة سبب معاناة بعض الأطفال المصابين بحالة نادرة تُعرف بمتلازمة ألان–هيرندون–دادلي من صعوبات حركية وتعلّمية عميقة. من خلال كشف كيفية حجب تغييرات جينية محددة وصول هرمون الغدة الدرقية إلى الدماغ، يسلط الباحثون الضوء على الكيمياء الخفية لتوصيف الشبكات الدماغية المبكرة ويشيرون إلى طرق جديدة قد تُستغل لتصميم علاجات مستقبلية.
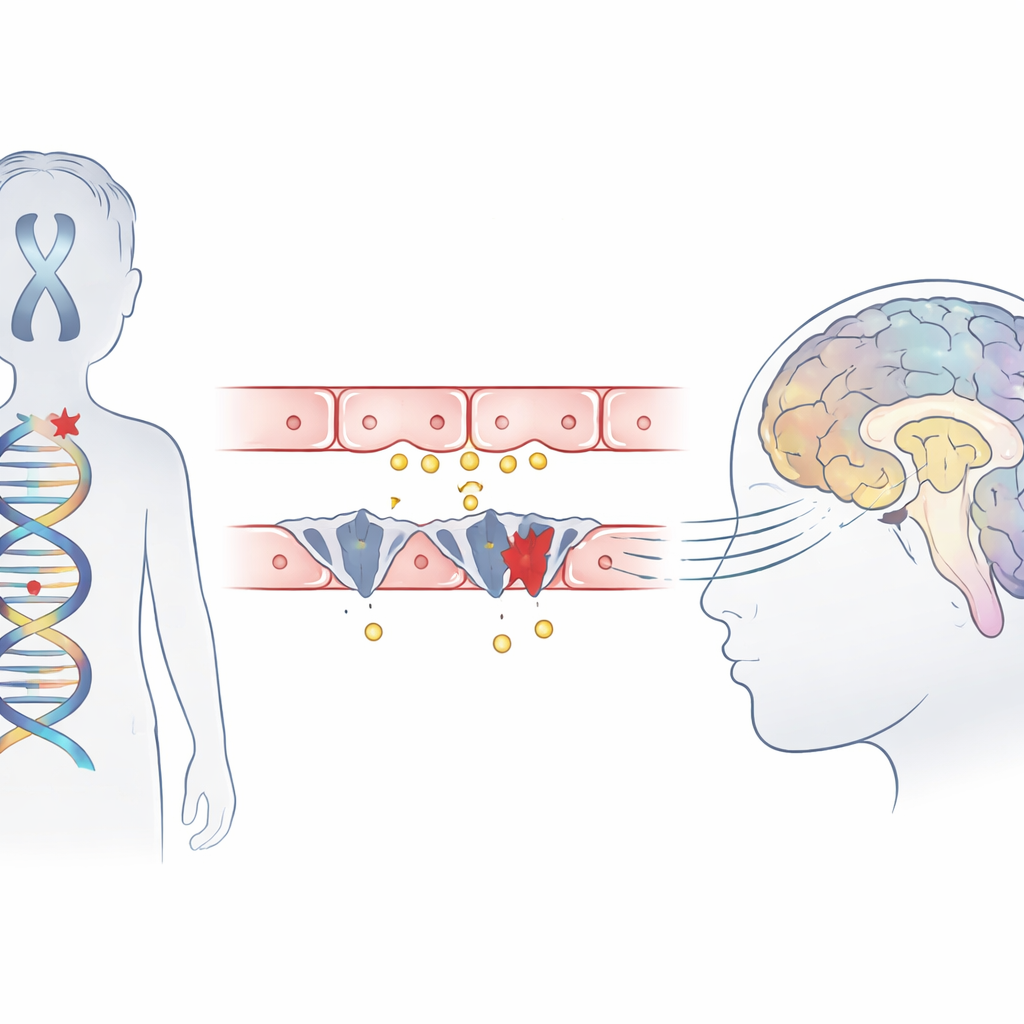
اضطراب طفولي نادر تحت المجهر
متلازمة ألان–هيرندون–دادلي هي حالة وراثية تصيب الذكور بشكل رئيسي وتؤدي إلى إعاقة عقلية شديدة، وعدم القدرة على الجلوس أو المشي بمفردهم، وتأخر توصيل الدوائر الدماغية كما يظهر في صور الرنين المغناطيسي. رغم أن فحوصات الدم تظهر نمطاً غير عادي لهرمونات الغدة الدرقية، فإن الغدة نفسها ليست السبب الرئيسي. بدلاً من ذلك، أشارت أعمال سابقة إلى أن بروتين ناقل يُسمى MCT8، الذي ينقل عادة الهرمون النشط إلى خلايا الدماغ، مفقود أو معطّل. تُبلغ هذه الدراسة عن ثلاثة أولاد من عائلات صينية غير مرتبطة أظهروا جميعاً الأعراض الكلاسيكية للمتلازمة وتستكشف بالضبط كيف تتداخل طفراتهم مع دخول الهرمون إلى الدماغ.
جينات تغلق بوابة الهرمون
باستخدام تسلسل الإكسوم، بحث الفريق في الحمض النووي للأطفال وحدد تغييرات ضارة في جين واحد، SLC16A2، الذي يشفر ناقل MCT8. حمل اثنان من الأولاد طفرات ‘‘قاصرة’’ جديدة تقصّر البروتين، أما الثالث فكان لديه طفرة في موقع تشابك حاسم حيث تُلتحم مقاطع الجين عادةً. أشارت نماذج الحاسوب لبنية البروتين إلى أن أحد هذه التغييرات يقطع جزءاً من قطعة حلزونية حرجة تُساعد في تشكيل جيب ارتباط الهرمون، مما يمنع الناقل من العمل. كانت جميع المتغيرات الثلاثة غائبة عن قواعد بيانات سكانية كبيرة وموجودة في أجزاء من الجين محفوظة بقوة عبر أنواع متعددة، مما يعزز احتمال كونها ممرِّضة بالفعل.
إشارات في خلايا الدم تكشف عن دماغ جائع
لمعرفة كيف تتجسّد هذه الطفرات في خلايا حية، قاس الباحثون نشاط الجينات في خلايا دم من الأطفال ووالديهم وأشخاص أصحاء كضوابط. كان تعبير جين SLC16A2 منخفضاً جداً لدى الأولاد المتأثرين، وهو ما يتوافق مع تحلل البروتين المعيب. في الوقت نفسه، أظهرت جينات أخرى تستجيب لمستويات الهرمون المحلية نمطاً مميزاً: ارتفعت DIO2 وHR، اللتان غالباً ما تزدادان عندما تشعر الخلايا بنقص في الهرمون، بينما انخفضت Nrgn وKIF9 بدرجة حادة؛ وهما يساعدان في توجيه نمو الأعصاب وتشكيل التشابكات وتغليف المحاور العصبية. هذه التغيرات مجتمعة ترسم صورة أن الأنسجة تحاول التعويض عن دخول غير كافٍ للهرمون، لكنها تفشل في دعم نمو دماغي طبيعي.
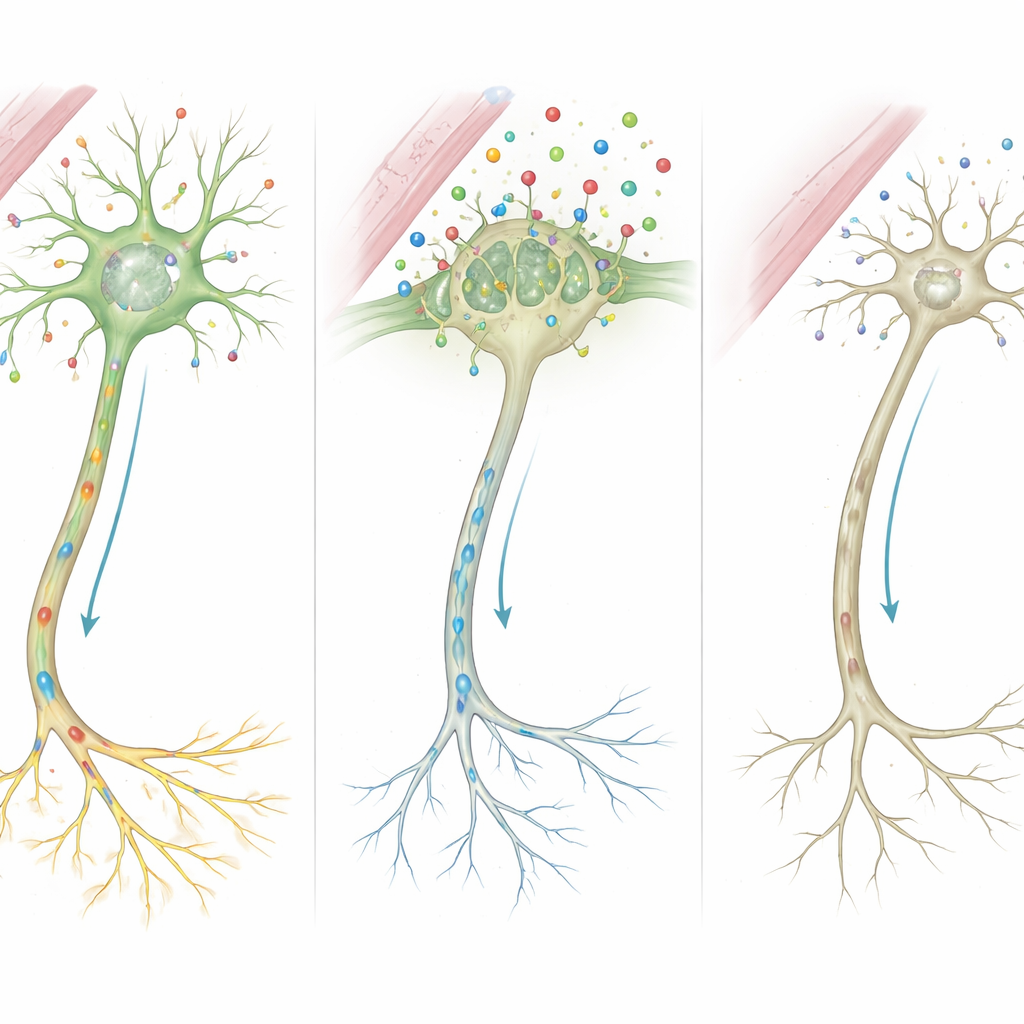
من الجزيئات إلى الدوائر المشوشة
بجمع هذه الأدلة، يقترح المؤلفون سلسلة مفصلة من الأحداث. بما أن MCT8 مفقود أو مقصّر، يعبر قدر أقل بكثير من هرمون الغدة الدرقية إلى خلايا الدماغ خلال فترات نمو حاسمة. محرومة من هذه الإشارة، لا يستطيع مركز التحكم الخلوي تشغيل الجينات التي تشكل بنية ألياف الأعصاب وقوة التشابكات بشكل صحيح. تعمل الخلايا الداعمة التي تغلف الأعصاب بالميلين، وتلك التي تعيد تدوير الناقلات الكيميائية، بشكل ضعيف أيضاً. مع مرور الوقت تتراكم هذه العيوب المجهرية لتظهر في الأعراض المرئية لدى المرضى: تأخر التميلين في الرنين المغناطيسي، وضعف أو تيبس العضلات، اضطرابات الحركة، وإعاقة إدراكية شديدة.
بناء منصة للعلاجات المستقبلية
بعيداً عن وصف ثلاث طفرات جديدة، تؤكد الدراسة صلة واضحة: كلما تضرر جين SLC16A2 بشدة أكبر، كانت الإعاقات أعمق. كما تؤسس مجموعة أدوات للعمل المستقبلي، بما في ذلك خطوط خلايا جذعية مشتقة من المرضى يمكن تحويلها إلى خلايا عصبية في المختبر. للغير متخصصين، الخلاصة الأساسية هي أن الدماغ يعتمد على توصيل في الوقت المناسب لهرمون الغدة الدرقية عبر بوابة جزيئية محددة. عندما تفشل تلك البوابة، يمكن أن يتعرقل التطور بشكل دائم. إن فهم هذا المسار بهذه العمق يبعث على الأمل بأن العلاجات يوماً ما — ربما عبر استهداف حاجز الدماغ-الدم، تعزيز ناقلات بديلة، أو تصحيح الجين نفسه — قد تستعيد على الأقل جزءاً من هذا الطريق الحيوي للهرمون.
الاستشهاد: Sun, X., Wang, C., Lin, L. et al. Novel SLC16A2 mutations impair thyroid hormone transport and drive neurodevelopmental deficits in Chinese patients with allan-herndon-dudley syndrome. Sci Rep 16, 11476 (2026). https://doi.org/10.1038/s41598-026-40703-3
الكلمات المفتاحية: متلازمة ألان-هيرندون-دادلي, نقل هرمون الغدة الدرقية, نقص MCT8, اضطرابات النمو العصبي, طفرات SLC16A2