Clear Sky Science · sv
En konformationsbenchmark för förutsägelse av optiska egenskaper med lösningsmedelsmedvetna grafneuronätverk
Varför det är viktigt att förutsäga molekylfärger
Från de ljusstarka pixlarna i telefoners skärmar till färgämnen i solceller och de lysande sonder som används för att se in i levande vävnad—många moderna teknologier bygger på molekyler som absorberar och avger ljus vid precis rätt färger. Att utforma dessa molekyler är svårt: små förändringar i struktur eller lösningsmedel kan förskjuta deras färger dramatiskt, och traditionella kvantkemiska beräkningar är för långsamma för att vägleda sökningar i stor skala. Denna artikel introducerar en ny datamängd och maskininlärningsmodeller som behandlar molekyler i full tredimension och explicit tar hänsyn till deras omgivande vätskemiljö, vilket möjliggör mycket snabbare och mer träffsäkra förutsägelser av optiska egenskaper.
Bygga en bättre karta över färgrika molekyler
Författarna samlade först in och rensade en stor samling experimentella data om hur organiska ”kromoforer”—de ljusabsorberande delarna av molekyler—beter sig i olika lösningsmedel. De kombinerade flera offentliga dataset och rättade noggrant problem som ogiltiga strukturbeskrivningar, inkonsekventa laddningstillstånd och missvisande strukturer som innehöll metaller. Resultatet är nablaColors, som täcker 13 731 unika molekyler och 26 369 kromofor–lösningsmedel-par med uppmätta absorptionsdata, och för många även emissionsvåglängder och ljusemissionseffektivitet (fotoluminescenskvantutbyte). Denna omsorgsfulla curering minskar brus som kan förvirra maskininlärningsmodeller och ger en pålitlig grund för vidare studier.
Lägga till den saknade tredje dimensionen
De flesta befintliga maskininlärningsverktyg för att förutsäga optiska egenskaper representerar molekyler som platta grafer: atomer är noder och kemiska bindningar är linjer. Exciterade tillstånd och ljusabsorption beror dock känsligt på verkliga tredimensionella former—bindningsvinklar, vridningar och svaga interaktioner—vilket dessa 2D-bilder inte kan fånga fullt ut. För att råda bot på detta genererade teamet 3D-strukturer för varje kromofor med en flerstegs pipeline: en initial grov 3D-layout, en snabbare semiempirisk kvantmetod och sedan mer precisa optimeringar med densitetsfunktionalteori (DFT), både i vakuum och med en implicit modell för det omgivande lösningsmedlet. Denna nya 3D-förlängning, nablaColors-3D, tillhandahåller flera konformationer per molekyl, var och en återspeglande olika nivåer av fysisk realism och beräkningskostnad. 
Lära neurala nät att se form och lösningsmedel
Med nablaColors-3D i handen byggde författarna en benchmark för att jämföra en rad maskininlärningsmodeller, från etablerade 2D-grafneuronätverk till toppmoderna 3D-arkitekturer som respekterar fysiska symmetrier i rummet. De designade också en ”lösningsmedelsmedveten” uppgradering: ett separat, lättviktigt neuralt nät kodar lösningsmedlets struktur från dess egen molekylära representation och producerar ett kompakt lösningsmedelsfingeravtryck. Detta fingeravtryck kombineras med kromoforens 3D-representation så att huvudmodellen kan lära sig hur vätskemiljön subtilt förskjuter molekylens geometri och elektroniska struktur. Genom att använda en rigorös scaffold-baserad datauppdelning säkerställer benchmarken att nära besläktade molekyler aldrig förekommer i både tränings- och testuppsättningar, så uppmätt prestanda speglar verklig generalisering snarare än memorering.
Hur mycket geometridetalj räcker?
En viktig praktisk fråga är om det är värt den höga beräkningskostnaden för mycket precisa geometrier. Teamet varierade systematiskt vilken typ av 3D-konformationer som gavs till varje modell—från billigare semiempiriska strukturer till mer krävande DFT-optimeringar i vakuum och i implicit lösningsmedel—samtidigt som alla träningsinställningar hölls fasta. Generellt förbättrade bättre geometrier förutsägelser, men effekten berodde på modellen och på om explicita lösningsmedelsfingeravtryck användes. När lösningsmedelsinbäddningar inkluderades krympte prestandaskillnaderna mellan geometridekällor, vilket visar att mycket av lösningsmedlets påverkan kunde fångas av denna separata kodning snarare än genom allt dyrare konformatorsberäkningar. För deras bästa modell visade de till och med att billiga strukturer genererade av standardkemisk programvara kunde ersätta kvantoptimerade sådana under träning med nästan ingen noggrannhetsförlust. 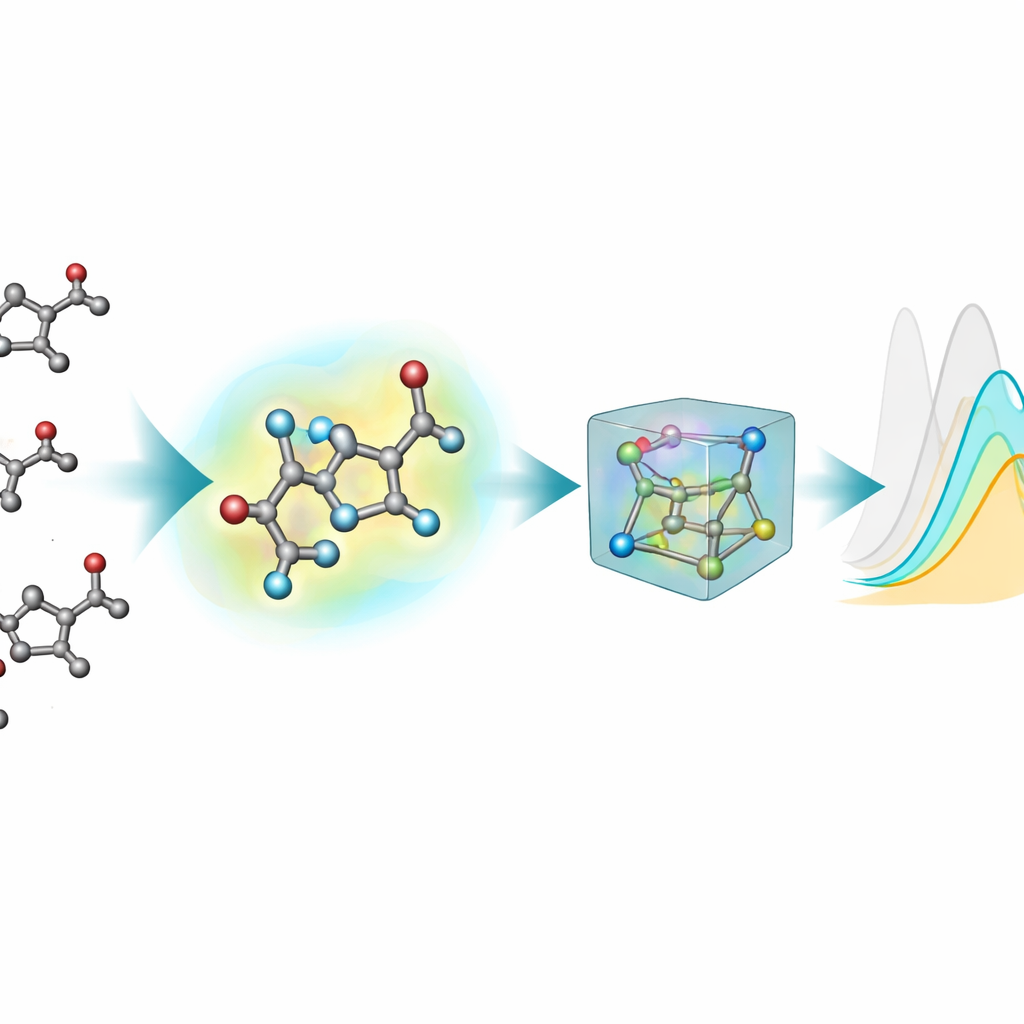
Ett språng bortom traditionella metoder
Bland alla testade modeller presterade en 3D-transformerbaserad arkitektur kallad UniMol+—utökad med lösningsmedelsinbäddningar i en variant som författarna kallar UniProp—bäst. UniProp uppnådde ett medelfel i absoluta termer på ungefär 16 nanometer för absorptionsvåglängder på ett hållet testset, mer än 30 % förbättring jämfört med starkaste 2D-baslinjen och betydligt bättre än en ofta använd tidsberoende DFT-metod, som hade ett fel på omkring 62 nanometer. Avgörande var att UniMol+ hade förtränats på stora kvantkemiska datamängder för att lära sig hur man förfinar grova 3D-strukturer mot högre nivåers geometrier. Denna förmåga till "geometribrusavstörning" gör att modellen kan acceptera relativt billiga konformerer vid prediktionstidpunkt medan den ändå fångar de fina strukturdetaljer som påverkar optiskt beteende.
Mot ett universellt verktyg för optisk design
Slutligen utvidgade författarna UniProp för att förutsäga inte bara absorptionspeakar utan även emissionsvåglängder och ljusemissionseffektivitet i en enda multitarget-modell. Den bibehöll hög noggrannhet över alla tre egenskaper, med bara en liten avvägning för absorption, vilket visar att samma 3D-funktioner fångar gemensamma fysikaliska faktorer bakom olika fotofysiska processer. För icke-specialister är huvudbudskapet att tredimensionella, lösningsmedelsmedvetna neurala nätverk—tränade på en noggrant curerad benchmark—nu kan överträffa traditionella kvantmetoder samtidigt som de körs ordningsstorlekar snabbare. Detta gör det realistiskt att virtuellt screena enorma bibliotek av kandidatfärgämnen, OLED-emittorer och fluorescerande sonder, och påskynda upptäckten av molekyler med exakt avstämda färger och ljusstyrka.
Citering: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Nyckelord: molekylär optik, grafneuronätverk, maskininlärningskemi, fluorescerande färgämnen, lösningsmedelseffekter