Clear Sky Science · pt
Um benchmark conformacional para previsão de propriedades ópticas com redes neurais gráficas conscientes do solvente
Por que prever as cores das moléculas importa
Dos pixels brilhantes nas telas de telefone aos corantes em células solares e às sondas luminosas usadas para visualizar tecidos vivos, muitas tecnologias modernas dependem de moléculas que absorvem e emitem luz nas cores certas. Projetar essas moléculas é difícil: pequenas mudanças na estrutura ou no solvente podem deslocar suas cores dramaticamente, e cálculos tradicionais de química quântica são lentos demais para orientar buscas em grande escala. Este trabalho introduz um novo conjunto de dados e modelos de aprendizado de máquina que tratam as moléculas em toda a sua tridimensionalidade e levam explicitamente em conta o ambiente líquido ao redor, possibilitando previsões de propriedades ópticas muito mais rápidas e precisas.
Construindo um mapa melhor de moléculas coloridas
Os autores primeiro reuniram e limp deram uma grande coleção de dados experimentais sobre como cromóforos orgânicos — as partes das moléculas que absorvem luz — se comportam em diferentes solventes. Eles combinaram vários conjuntos de dados públicos e então corrigiram minuciosamente problemas como descrições estruturais inválidas, estados de carga inconsistentes e estruturas enganosas contendo metais. O resultado é o nablaColors, cobrindo 13.731 moléculas únicas e 26.369 pares cromóforo–solvente com absorção medida, e para muitas delas também comprimentos de onda de emissão e eficiência de emissão de luz (rendimento quântico de fotoluminescência). Essa curadoria cuidadosa reduz o ruído que pode confundir modelos de aprendizado de máquina e estabelece uma base confiável para estudos futuros.
Adicionando a terceira dimensão que faltava
A maioria das ferramentas de aprendizado de máquina existentes para prever propriedades ópticas representa moléculas como grafos planos: átomos como nós e ligações químicas como linhas. Contudo, estados excitados e absorção de luz dependem sensivelmente das formas tridimensionais reais — ângulos de ligação, torções e interações fracas — que essas imagens 2D não capturam completamente. Para remediar isso, a equipe gerou estruturas 3D para cada cromóforo usando um pipeline em múltiplas etapas: um layout 3D inicial aproximado, um método quântico semi-empírico mais rápido e, em seguida, otimizações mais precisas por teoria do funcional da densidade (DFT), tanto em vácuo quanto com um modelo implícito do solvente circundante. Esta nova extensão 3D, nablaColors-3D, fornece múltiplas conformações por molécula, cada uma refletindo um nível diferente de realismo físico e custo computacional. 
Ensinando redes neurais a ver forma e solvente
Com o nablaColors-3D em mãos, os autores construíram um benchmark para comparar uma gama de modelos de aprendizado de máquina, desde redes neurais gráficas 2D estabelecidas até arquiteturas 3D de ponta que respeitam simetrias físicas no espaço. Eles também desenharam uma melhora “consciente do solvente”: uma rede neural separada e leve codifica a estrutura do solvente a partir de sua própria representação molecular, produzindo uma impressão digital compacta do solvente. Essa impressão é combinada com a representação 3D do cromóforo para que o modelo principal aprenda como o ambiente líquido desloca sutilmente a geometria e a estrutura eletrônica da molécula. Ao usar uma divisão de dados rigorosa baseada em scaffold, o benchmark garante que moléculas estreitamente relacionadas nunca apareçam tanto no conjunto de treino quanto no de teste, de modo que o desempenho medido reflita generalização verdadeira em vez de memorização.
Quanta detalhe geométrico é suficiente?
Uma questão prática importante é se vale a pena pagar o alto custo computacional de geometrias muito precisas. A equipe variou sistematicamente o tipo de conformações 3D fornecidas a cada modelo — desde estruturas semi-empíricas mais baratas até otimizações DFT mais exigentes em vácuo e em solvente implícito — mantendo todas as configurações de treino fixas. Em geral, geometrias melhores tenderam a melhorar as previsões, mas o efeito dependia do modelo e de o quanto impressões digitais explícitas do solvente foram usadas. Uma vez incluídas as incorporações do solvente, as diferenças de desempenho entre as fontes de geometria diminuíram, mostrando que grande parte da influência do solvente pode ser capturada por essa codificação separada em vez de por cálculos cada vez mais caros de conformadores. Para seu melhor modelo, eles mostraram inclusive que estruturas baratas geradas por softwares químicos padrão poderiam substituir as otimizadas por métodos quânticos durante o treinamento com quase nenhuma perda de precisão. 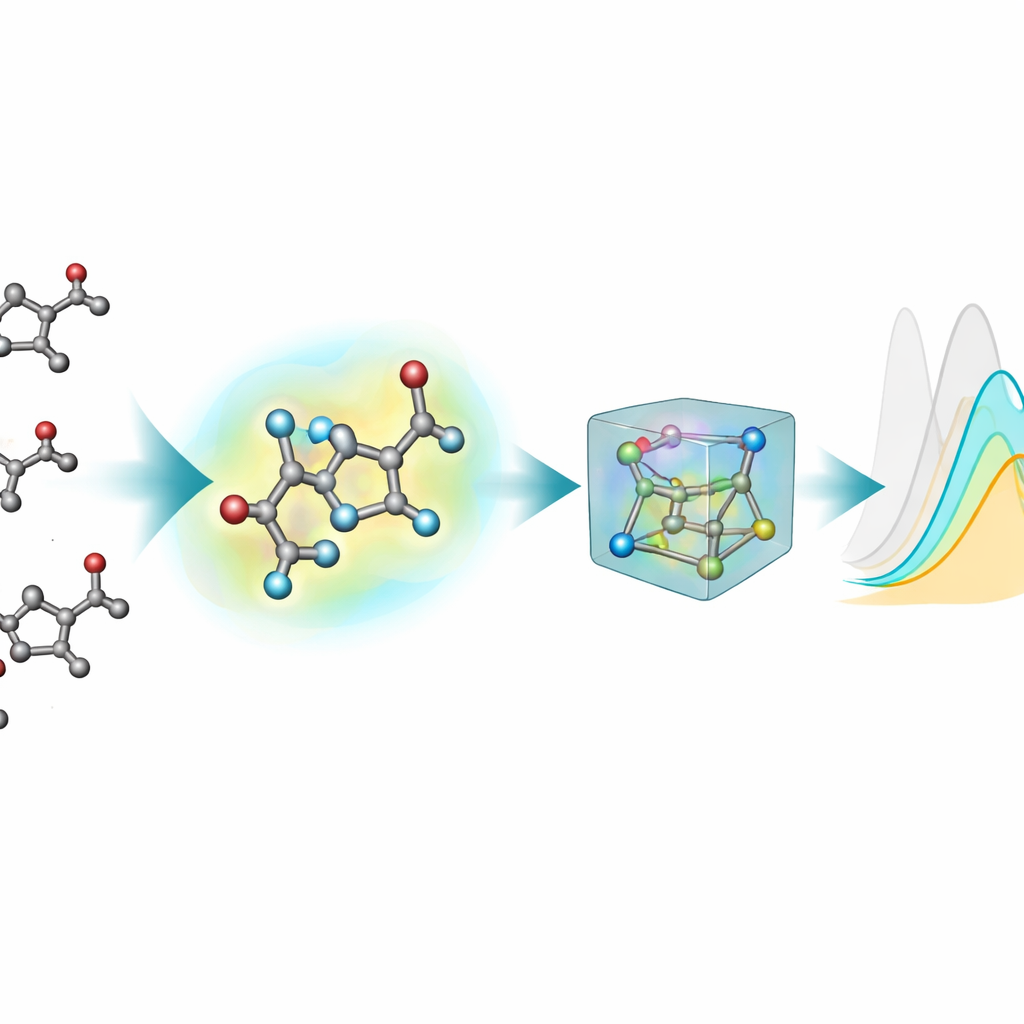
Um salto além dos métodos tradicionais
Entre todos os modelos testados, uma arquitetura 3D baseada em transformer chamada UniMol+ — aumentada com incorporações de solvente em uma variante que os autores chamam UniProp — teve o melhor desempenho. A UniProp alcançou um erro absoluto médio de cerca de 16 nanômetros para comprimentos de onda de absorção em um conjunto de teste mantido à parte, mais de 30% de melhoria sobre a melhor linha de base 2D e muito superior a um método amplamente usado de DFT dependente do tempo, que errou por cerca de 62 nanômetros. Crucialmente, o UniMol+ havia sido pré-treinado em grandes conjuntos de dados de química quântica para aprender a refinar estruturas 3D aproximadas rumo a geometrias de alto nível. Essa habilidade de “desruído geométrico” permite que ele aceite conformadores relativamente baratos na hora da predição enquanto ainda captura os detalhes estruturais finos que importam para o comportamento óptico.
Rumo a uma ferramenta universal de design óptico
Por fim, os autores estenderam o UniProp para prever não apenas picos de absorção, mas também comprimentos de onda de emissão e eficiência de emissão de luz em um único modelo multitarefa. Ele manteve alta precisão nas três propriedades, com apenas uma ligeira troca para absorção, demonstrando que as mesmas características 3D capturam fatores físicos compartilhados por diferentes processos fotofísicos. Para não especialistas, a principal conclusão é que redes neurais tridimensionais e conscientes do solvente — treinadas em um benchmark cuidadosamente curado — agora podem superar métodos quânticos tradicionais enquanto rodam ordens de magnitude mais rápido. Isso torna realista a triagem virtual de enormes bibliotecas de corantes candidatos, emissores para OLEDs e sondas fluorescentes, acelerando a descoberta de moléculas com cores e brilho precisamente ajustados.
Citação: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Palavras-chave: óptica molecular, redes neurais gráficas, química por aprendizado de máquina, corantes fluorescentes, efeitos do solvente