Clear Sky Science · de
Ein konformationelles Benchmark für die Vorhersage optischer Eigenschaften mit lösungsmittelbewussten Graph-Neuronalen Netzen
Warum die Vorhersage von Molekülfarben wichtig ist
Von den leuchtenden Pixeln in Smartphone-Displays über Farbstoffe in Solarzellen bis zu den fluoreszierenden Sonden, mit denen man in lebendes Gewebe hineinsieht: zahlreiche moderne Technologien beruhen auf Molekülen, die Licht in genau den richtigen Farben absorbieren und emittieren. Diese Moleküle zu entwerfen ist schwierig: schon kleine Änderungen in der Struktur oder im Lösungsmittel können die Farbe stark verschieben, und klassische quantenchemische Berechnungen sind zu langsam, um groß angelegte Suchläufe zu steuern. Diese Arbeit stellt einen neuen Datensatz und maschinelle Lernmodelle vor, die Moleküle in voller dreidimensionaler Gestalt behandeln und deren umgebende Flüssigkeit explizit berücksichtigen, was deutlich schnellere und genauere Vorhersagen optischer Eigenschaften ermöglicht.
Eine bessere Karte farbiger Moleküle erstellen
Die Autoren haben zunächst eine große Sammlung experimenteller Daten darüber zusammengestellt und bereinigt, wie organische „Chromophore“—die lichtabsorbierenden Teile von Molekülen—sich in verschiedenen Lösungsmitteln verhalten. Sie kombinierten mehrere öffentliche Datensätze und korrigierten sorgfältig Probleme wie ungültige Strukturangaben, inkonsistente Ladungszustände und irreführende metallhaltige Strukturen. Das Ergebnis ist nablaColors, das 13.731 eindeutige Moleküle und 26.369 Chromophor–Lösungsmittel-Paare mit gemessenen Absorptionsdaten abdeckt, und für viele davon auch Emissionswellenlängen und Lichtausbeuten (photolumineszente Quantenausbeute). Diese sorgfältige Kuration reduziert Rauschen, das maschinelle Lernmodelle verwirren kann, und schafft eine verlässliche Grundlage für weitere Studien.
Die fehlende dritte Dimension hinzufügen
Die meisten bestehenden Werkzeuge des maschinellen Lernens zur Vorhersage optischer Eigenschaften stellen Moleküle als flache Graphen dar: Atome sind Knoten und chemische Bindungen Linien. Anregungszustände und Lichtabsorption hängen jedoch empfindlich von realen dreidimensionalen Formen—Bindungswinkeln, Verdrehungen und schwachen Wechselwirkungen—ab, die diese 2D-Bilder nicht vollständig erfassen. Um dem abzuhelfen erzeugte das Team 3D-Strukturen für jedes Chromophor mithilfe einer mehrstufigen Pipeline: eine anfängliche grobe 3D-Anordnung, eine schnellere semiempirische Quantenmethode und dann genauere Dichtefunktionaltheorie-(DFT)-Optimierungen, sowohl im Vakuum als auch mit einem impliziten Modell des umgebenden Lösungsmittels. Diese neue 3D-Erweiterung, nablaColors-3D, stellt mehrere Konformationen pro Molekül bereit, die jeweils ein unterschiedliches Maß an physikalischer Realismus und Rechenaufwand widerspiegeln. 
Neuronale Netze das Sehen von Form und Lösungsmittel lehren
Mit nablaColors-3D bauten die Autoren ein Benchmark auf, um eine Reihe von ML-Modellen zu vergleichen, von etablierten 2D-Graphneuronalen Netzen bis zu modernen 3D-Architekturen, die physikalische Symmetrien im Raum respektieren. Sie entwarfen außerdem ein „lösungmittelbewusstes“ Upgrade: ein separates, leichtgewichtiges neuronales Netz kodiert die Struktur des Lösungsmittels aus seiner eigenen molekularen Darstellung und erzeugt so einen kompakten Lösungsmittelfingerprint. Dieser Fingerprint wird mit der 3D-Darstellung des Chromophors kombiniert, damit das Hauptmodell lernen kann, wie die flüssige Umgebung die Molekülgeometrie und elektronische Struktur subtil verschiebt. Durch eine strikte, auf Scaffold basierende Datenaufteilung stellt das Benchmark sicher, dass eng verwandte Moleküle niemals sowohl in Trainings- als auch in Testmengen vorkommen, sodass die gemessene Leistung echte Generalisierung statt Auswendiglernen widerspiegelt.
Wie viele Geometrie-Details sind genug?
Eine wichtige praktische Frage ist, ob sich die hohen Rechenkosten sehr genauer Geometrien lohnen. Das Team variierte systematisch die Art der 3D-Konformationen, die jedem Modell vorgelegt wurden—von günstigeren semiempirischen Strukturen bis hin zu aufwändigeren DFT-Optimierungen im Vakuum und im impliziten Lösungsmittel—während alle Trainingsparameter unverändert blieben. Allgemein verbesserten bessere Geometrien die Vorhersagen, doch der Effekt hing vom Modell und davon ab, ob explizite Lösungsmittelfingerprints verwendet wurden. Sobald Lösungsmittel-Embedding eingeschlossen war, schrumpften die Leistungsunterschiede zwischen den Geometriequellen, was zeigt, dass ein Großteil des Einflusses des Lösungsmittels durch diese separate Kodierung erfasst werden kann, statt durch immer teurere Konformer-Rechnungen. Für ihr bestes Modell zeigten sie sogar, dass kostengünstige Strukturen, die mit gängiger Chemiesoftware erzeugt wurden, die quantenchemisch optimierten Strukturen beim Training praktisch ohne Genauigkeitsverlust ersetzen können. 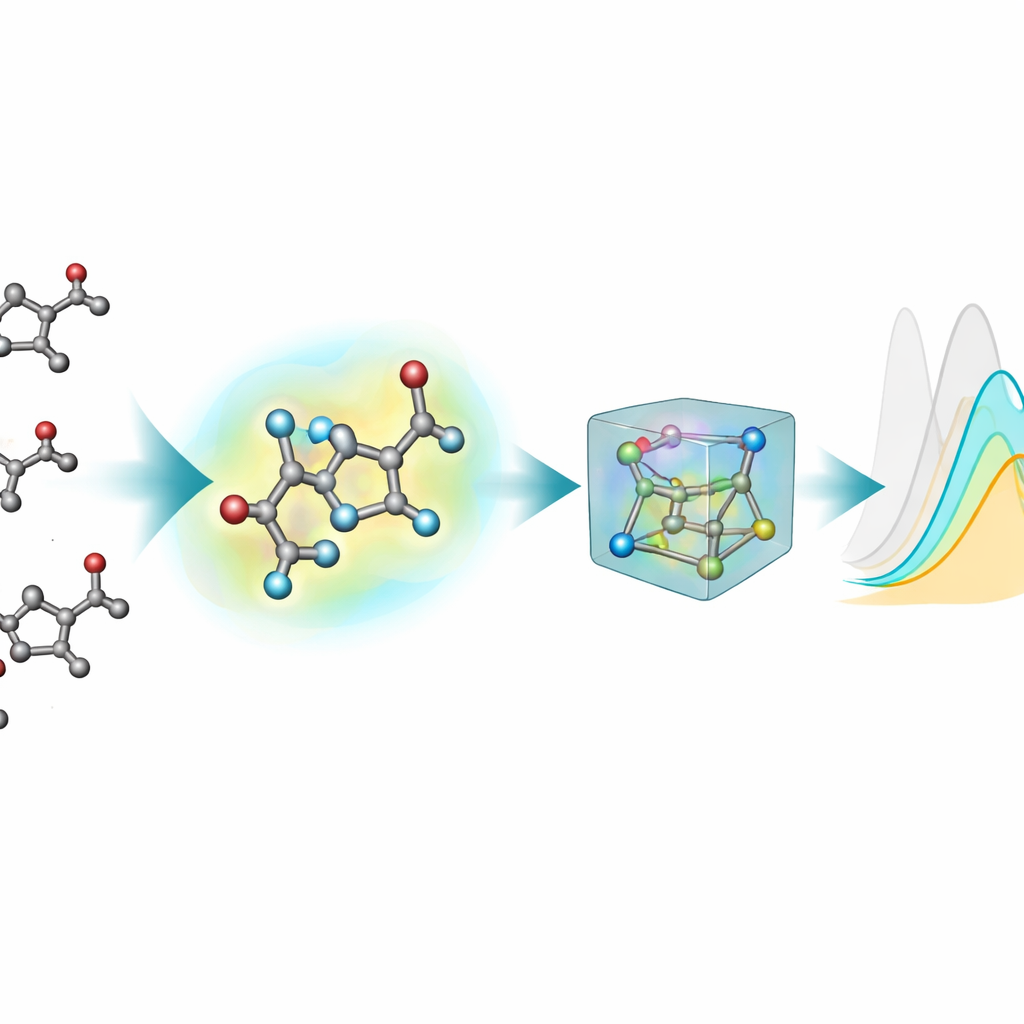
Ein Sprung über traditionelle Methoden hinaus
Unter allen getesteten Modellen schnitt eine 3D-Transformer-basierte Architektur namens UniMol+—ergänzt durch Lösungsmittel-Embeddings in einer Variante, die die Autoren UniProp nennen—am besten ab. UniProp erzielte auf einer zurückgehaltenen Testmenge einen mittleren absoluten Fehler von etwa 16 Nanometern für Absorptionswellenlängen, mehr als 30 % besser als die stärkste 2D-Baseline und deutlich besser als eine weit verbreitete zeitabhängige DFT-Methode, die um etwa 62 Nanometer danebenlag. Entscheidenderweise war UniMol+ vortrainiert worden auf großen quantenchemischen Datensätzen, um zu lernen, wie grobe 3D-Strukturen in richtung hochstufiger Geometrien verfeinert werden. Diese Fähigkeit zur „Geometrie-Entrauschung“ erlaubt es ihm, relativ günstige Konformer zur Vorhersage zu akzeptieren und trotzdem die feinen Strukturdetaillen zu erfassen, die für das optische Verhalten wichtig sind.
Richtung eines universellen Werkzeugs für optisches Design
Abschließend erweiterten die Autoren UniProp so, dass es nicht nur Absorptionspeaks vorhersagen kann, sondern auch Emissionswellenlängen und Lichtemissionseffizienz in einem einzigen Multizielmodell. Es behielt hohe Genauigkeit über alle drei Eigenschaften hinweg bei, mit nur einem geringen Kompromiss bei der Absorption, und zeigte damit, dass dieselben 3D-Merkmale gemeinsame physikalische Faktoren hinter verschiedenen photophysikalischen Prozessen erfassen. Für Nichtfachleute ist die wichtigste Erkenntnis: dreidimensionale, lösungsmittelbewusste neuronale Netze—trainiert auf einem sorgfältig kuratierten Benchmark—können nun traditionelle Quantenmethoden übertreffen und laufen dabei um viele Größenordnungen schneller. Das macht es realistisch, riesige Bibliotheken potenzieller Farbstoffe, OLED-Emitter und fluoreszierender Sonden virtuell zu screenen und die Entdeckung von Molekülen mit präzise abgestimmten Farben und Leuchtstärken zu beschleunigen.
Zitation: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Schlüsselwörter: molekulare Optik, graphneuronale Netze, maschinelles Lernen Chemie, fluoreszierende Farbstoffe, Lösungsmittel-Effekte