Clear Sky Science · fr
Un banc d'essai conformationnel pour la prédiction des propriétés optiques avec des réseaux de neurones graphiques sensibles au solvant
Pourquoi prédire la couleur des molécules importe
Des pixels vifs des écrans de téléphone aux colorants des cellules solaires en passant par les sondes luminescentes utilisées pour observer les tissus vivants, de nombreuses technologies modernes reposent sur des molécules qui absorbent et émettent la lumière à des couleurs précises. Concevoir ces molécules est difficile : de petits changements de structure ou de solvant peuvent décaler leurs couleurs de façon spectaculaire, et les calculs traditionnels de chimie quantique sont trop lents pour guider des recherches à grande échelle. Cet article présente un nouveau jeu de données et des modèles d'apprentissage automatique qui traitent les molécules en trois dimensions complètes et tiennent explicitement compte de leur environnement liquide, permettant des prédictions des propriétés optiques beaucoup plus rapides et plus précises.
Construire une meilleure cartographie des molécules colorées
Les auteurs ont d'abord rassemblé et nettoyé une grande collection de données expérimentales sur le comportement des « chromophores » organiques — les parties des molécules qui absorbent la lumière — dans différents solvants. Ils ont combiné plusieurs jeux de données publics puis corrigé minutieusement des problèmes tels que des descriptions structurelles invalides, des états de charge inconsistants et des structures trompeuses contenant des métaux. Le résultat est nablaColors, couvrant 13 731 molécules uniques et 26 369 paires chromophore–solvant avec des mesures d'absorption, et pour beaucoup d'entre elles des longueurs d'onde d'émission et des efficacités d'émission lumineuse (rendement quantique de photoluminescence). Cette curation soignée réduit le bruit susceptible de perturber les modèles d'apprentissage automatique et établit une base fiable pour des études ultérieures.
Ajouter la troisième dimension manquante
La plupart des outils d'apprentissage automatique existants pour prédire les propriétés optiques représentent les molécules comme des graphes plats : les atomes sont des nœuds et les liaisons chimiques des lignes. Or, les états excités et l'absorption lumineuse dépendent fortement des formes tridimensionnelles réelles — angles de liaisons, torsions et interactions faibles — que ces images 2D ne peuvent pas pleinement capturer. Pour remédier à cela, l'équipe a généré des structures 3D pour chaque chromophore via une chaîne de traitement en plusieurs étapes : une mise en place 3D initiale approximative, une méthode semi‑empirique quantique plus rapide, puis des optimisations plus précises en théorie de la fonctionnelle de la densité (DFT), à la fois en phase gazeuse et avec un modèle implicite du solvant environnant. Cette extension 3D, nablaColors-3D, fournit plusieurs conformations par molécule, chacune reflétant un niveau différent de réalisme physique et de coût de calcul. 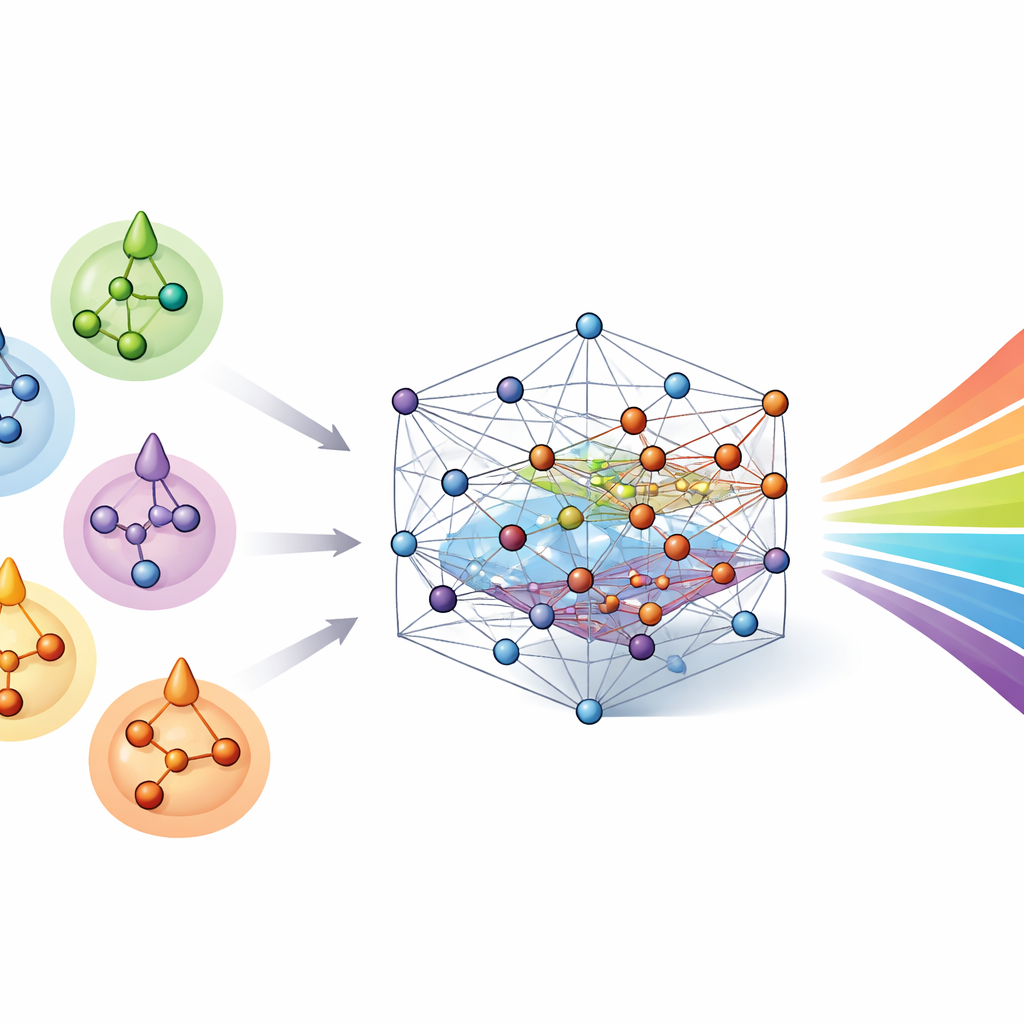
Apprendre aux réseaux neuronaux à voir la forme et le solvant
Avec nablaColors-3D en main, les auteurs ont construit un banc d'essai pour comparer une gamme de modèles d'apprentissage automatique, des réseaux de neurones graphiques 2D établis aux architectures 3D à la pointe qui respectent les symétries physiques de l'espace. Ils ont également conçu une amélioration « sensible au solvant » : un réseau neuronal séparé et léger code la structure du solvant à partir de sa propre représentation moléculaire, produisant une empreinte compacte du solvant. Cette empreinte est combinée à la représentation 3D du chromophore afin que le modèle principal puisse apprendre comment l'environnement liquide déplace subtilement la géométrie et la structure électronique de la molécule. En utilisant une séparation rigoureuse des données basée sur le squelette moléculaire (scaffold), le banc d'essai garantit que des molécules étroitement apparentées n'apparaissent jamais à la fois dans les ensembles d'entraînement et de test, de sorte que la performance mesurée reflète une véritable généralisation plutôt qu'une mémorisation.
Quelle précision géométrique est suffisante ?
Une question pratique importante est de savoir s'il vaut la peine de payer le coût computationnel élevé de géométries très précises. L'équipe a fait varier systématiquement le type de conformations 3D fournies à chaque modèle — allant de structures semi‑empiriques moins coûteuses à des optimisations DFT plus exigeantes en phase gazeuse et en solvant implicite — tout en gardant tous les réglages d'entraînement constants. En général, de meilleures géométries tende nt à améliorer les prédictions, mais l'effet dépendait du modèle et de l'utilisation ou non d'empreintes explicites du solvant. Une fois les embeddings du solvant inclus, les différences de performance entre les sources de géométrie se sont réduites, montrant qu'une grande partie de l'influence du solvant pouvait être capturée par cet encodage séparé plutôt que par des calculs de conformères toujours plus coûteux. Pour leur meilleur modèle, ils ont même montré que des structures peu coûteuses générées par des logiciels chimiques standards pouvaient remplacer des structures optimisées quantiquement lors de l'entraînement avec presque aucune perte de précision. 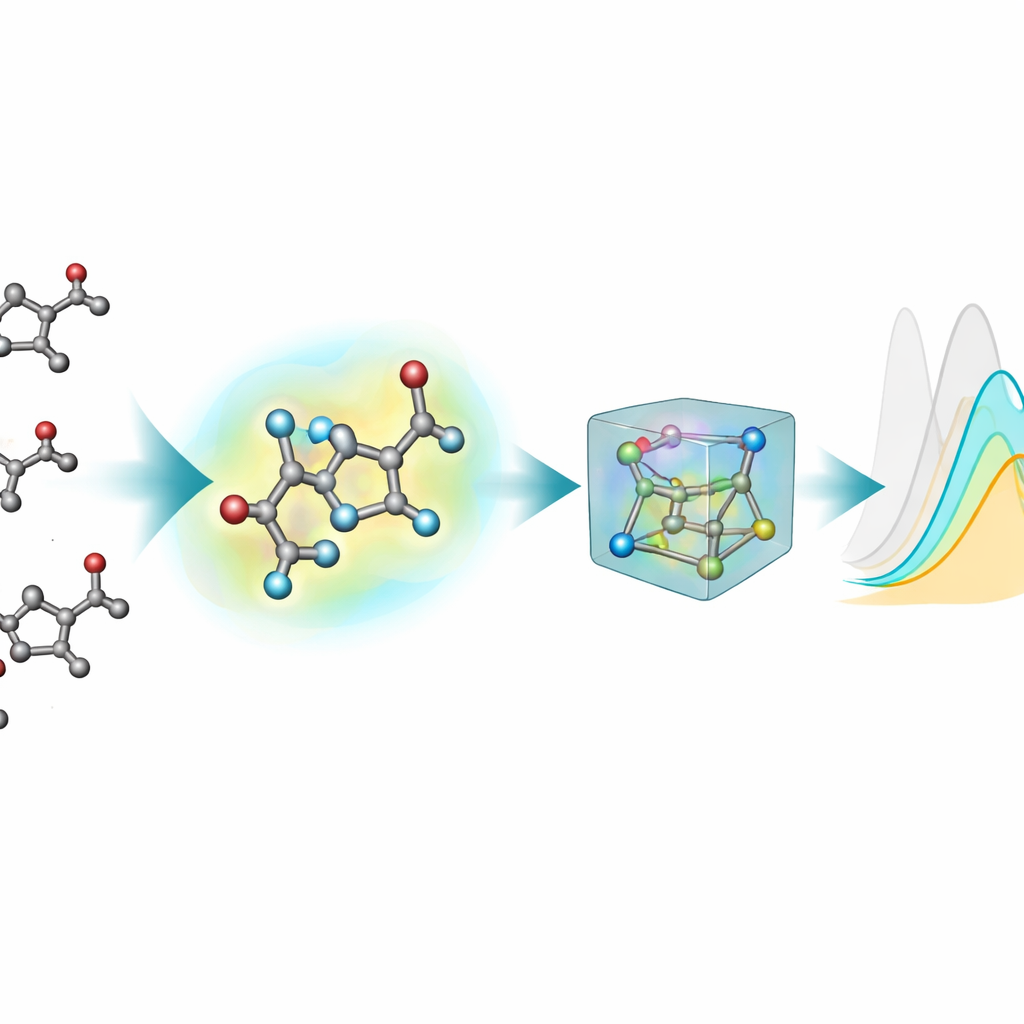
Un saut au‑delà des méthodes traditionnelles
Parmi tous les modèles testés, une architecture 3D basée sur un transformeur appelée UniMol+ — augmentée d'embeddings de solvant dans une variante que les auteurs nomment UniProp — a donné les meilleurs résultats. UniProp a atteint une erreur absolue moyenne d'environ 16 nanomètres pour les longueurs d'onde d'absorption sur un jeu de test tenu à part, soit une amélioration de plus de 30 % par rapport au meilleur modèle 2D et bien meilleure qu'une méthode TD‑DFT largement utilisée, qui commettait une erreur d'environ 62 nanomètres. Fait crucial, UniMol+ avait été préentraîné sur de grands jeux de données de chimie quantique pour apprendre à affiner des structures 3D approximatives vers des géométries de haut niveau. Cette capacité de « débruitage géométrique » lui permet d'accepter des conformères relativement bon marché au moment de la prédiction tout en capturant les détails structuraux fins qui importent pour le comportement optique.
Vers un outil universel de conception optique
Enfin, les auteurs ont étendu UniProp pour prédire non seulement les pics d'absorption, mais aussi les longueurs d'onde d'émission et l'efficacité d'émission lumineuse dans un modèle multitâche unique. Il a conservé une grande précision sur les trois propriétés, avec seulement un léger compromis pour l'absorption, démontrant que les mêmes caractéristiques 3D capturent des facteurs physiques communs à différents processus photophysiques. Pour les non‑spécialistes, l'idée principale est que des réseaux neuronaux tridimensionnels et sensibles au solvant — entraînés sur un banc d'essai soigneusement curaté — peuvent désormais dépasser les méthodes quantiques traditionnelles tout en s'exécutant des ordres de grandeur plus rapidement. Cela rend réaliste le criblage virtuel de vastes bibliothèques de colorants candidats, d'émetteurs OLED et de sondes fluorescentes, accélérant la découverte de molécules aux couleurs et à la luminosité précisément ajustées.
Citation: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Mots-clés: optique moléculaire, réseaux de neurones graphiques, apprentissage automatique en chimie, colorants fluorescents, effets du solvant