Clear Sky Science · nl
Een conformationele benchmark voor voorspelling van optische eigenschappen met oplosmiddelbewuste graf-neurale netwerken
Waarom het voorspellen van molecuulkleur ertoe doet
Van de heldere pixels in telefoonschermen tot de kleurstoffen in zonnecellen en de gloeiende sondes die gebruikt worden om in levend weefsel te kijken, veel moderne technologieën vertrouwen op moleculen die licht absorberen en uitzenden op precies de juiste kleuren. Het ontwerpen van zulke moleculen is moeilijk: kleine veranderingen in structuur of oplosmiddel kunnen hun kleur sterk verschuiven, en traditionele kwantumchemische berekeningen zijn te traag om grootschalige zoektochten te sturen. Dit artikel introduceert een nieuwe dataset en machine-learningmodellen die moleculen in volledige drie dimensies behandelen en expliciet rekening houden met hun omringende vloeibare omgeving, waardoor voorspellingen van optische eigenschappen veel sneller en nauwkeuriger worden.
Een betere kaart van gekleurde moleculen bouwen
De auteurs verzamelden en schonen eerst een grote collectie experimentele gegevens over hoe organische "chromoforen"—de licht-absorberende delen van moleculen—zich gedragen in verschillende oplosmiddelen. Ze combineerden meerdere openbare datasets en corrigeerden zorgvuldig problemen zoals ongeldige structurele beschrijvingen, inconsistente ladingsstaten en misleidende metaal-bevattende structuren. Het resultaat is nablaColors, met 13.731 unieke moleculen en 26.369 chromofoor–oplosmiddelkoppels met gemeten absorptie, en voor veel daarvan ook emissiegolflengten en lichtuitzendings-efficiëntie (photoluminescentie-kwantumopbrengst). Deze zorgvuldige curatie vermindert ruis die machine-learningmodellen kan verwarren en legt een betrouwbare basis voor verder onderzoek.
De ontbrekende derde dimensie toevoegen
De meeste bestaande machine-learninghulpmiddelen voor het voorspellen van optische eigenschappen stellen moleculen voor als platte grafen: atomen zijn knooppunten en bindingen lijnen. Geëxciteerde toestanden en lichtabsorptie hangen echter gevoelig af van echte driedimensionale vormen—bindingshoeken, verdraaiingen en zwakke interacties—die deze 2D-voorstellingen niet volledig kunnen vangen. Om dit te verhelpen genereerde het team 3D-structuren voor ieder chromofoor met een meerstaps-pijplijn: een aanvankelijke grove 3D-indeling, een snellere semi-empirische kwantummethode, en vervolgens nauwkeurigere dichtheidsfunctionaaltheorie (DFT)-optimalisaties, zowel in vacuüm als met een impliciet model van het omringende oplosmiddel. Deze nieuwe 3D-uitbreiding, nablaColors-3D, biedt meerdere conformaties per molecuul, elk reflecterend een ander niveau van fysieke realisme en computationele kost. 
Neurale netwerken leren vorm en oplosmiddel te zien
Met nablaColors-3D bouwden de auteurs een benchmark om een reeks machine-learningmodellen te vergelijken, van gevestigde 2D-graf-neurale netwerken tot state-of-the-art 3D-architecturen die fysieke symmetrieën in de ruimte respecteren. Ze ontwierpen ook een "oplosmiddelbewuste" upgrade: een apart, lichtgewicht neurale netwerk codeert de structuur van het oplosmiddel vanuit zijn eigen moleculaire representatie en produceert een compacte oplosmiddeleigenschap (fingerprint). Deze fingerprint wordt gecombineerd met de 3D-representatie van het chromofoor zodat het hoofdmodel kan leren hoe de vloeibare omgeving subtiel de geometrie en elektronische structuur van het molecuul verschuift. Door een rigoureuze scaffold-gebaseerde datasplitsing te gebruiken, zorgt de benchmark ervoor dat nauw verwante moleculen nooit zowel in de trainings- als in de testset voorkomen, zodat gemeten prestatie echte generalisatie weerspiegelt in plaats van memorisatie.
Hoeveel detaillering van geometrie is genoeg?
Een belangrijke praktische vraag is of het de hoge computationele prijs van zeer nauwkeurige geometrieën waard is. Het team varieerde systematisch het type 3D-conformaties dat aan elk model werd gegeven—lopend van goedkopere semi-empirische structuren tot zwaardere DFT-optimalisaties in vacuüm en in impliciet oplosmiddel—terwijl alle trainingsinstellingen gelijk werden gehouden. Over het algemeen verbeterden betere geometrieën de voorspellingen, maar het effect hing af van het model en van het gebruik van expliciete oplosmiddelembeddings. Zodra oplosmiddelembeddings waren opgenomen, werden prestatieverschillen tussen geometriebronnen kleiner, wat aantoont dat een groot deel van de invloed van het oplosmiddel kan worden vastgelegd door deze afzonderlijke codering in plaats van door steeds duurdere conformatie-berekeningen. Voor hun beste model toonden ze zelfs aan dat goedkope structuren gegenereerd door standaard chemiesoftware tijdens training kwantum-geoptimaliseerde structuren konden vervangen met bijna geen nauwkeurigheidsverlies. 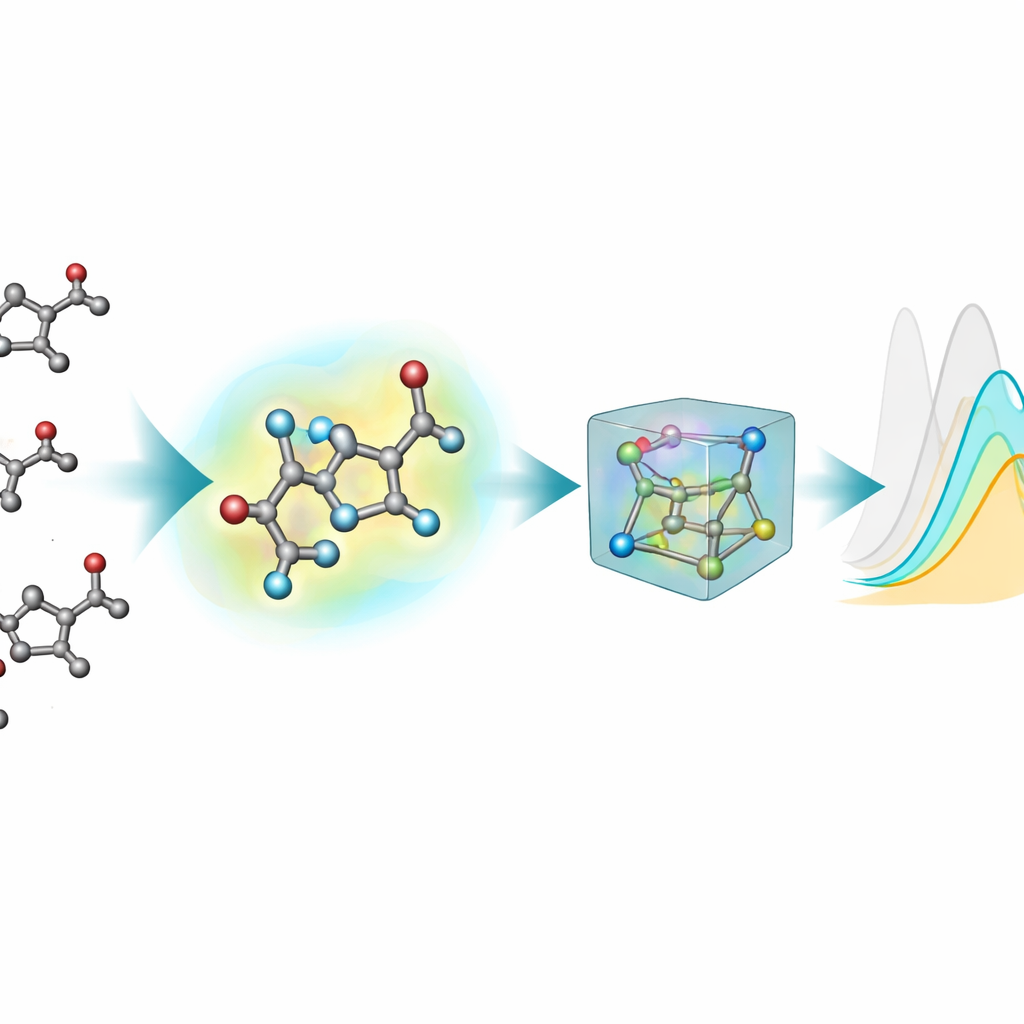
Een sprong voorbij traditionele methoden
Van alle geteste modellen presteerde een 3D-transformer-gebaseerde architectuur genaamd UniMol+—aangevuld met oplosmiddelembeddings in een variant die de auteurs UniProp noemen—het best. UniProp behaalde een gemiddelde absolute fout van ongeveer 16 nanometer voor absorptiegolflengten op een achtergehouden testset, meer dan 30% verbetering ten opzichte van de sterkste 2D-baseline en aanzienlijk beter dan een veelgebruikte tijdsafhankelijke DFT-methode, die er ongeveer 62 nanometer naast zat. Cruciaal is dat UniMol+ vooraf was voorgetraind op grote kwantumchemie-datasets om te leren hoe grove 3D-structuren naar hoogwaardige geometrieën verfijnd kunnen worden. Deze "geometrie-ruisonderdrukking"-vaardigheid laat het toe relatief goedkope conformaties te accepteren tijdens voorspellingstijd terwijl het toch de fijne structurele details vastlegt die van belang zijn voor optisch gedrag.
Naar een universeel ontwerpgereedschap voor optica
Tenslotte breidden de auteurs UniProp uit om niet alleen absorptiepieken te voorspellen, maar ook emissiegolflengten en lichtuitzendings-efficiëntie in één multitarget-model. Het behield een hoge nauwkeurigheid over alle drie eigenschappen, met slechts een lichte afweging voor absorptie, en toonde aan dat dezelfde 3D-kenmerken gedeelde fysieke factoren achter verschillende fotofysische processen vangen. Voor niet-specialisten is de belangrijkste conclusie dat driedimensionale, oplosmiddelbewuste neurale netwerken—getraind op een zorgvuldig samengestelde benchmark—nu traditionele kwantummethoden kunnen overtreffen terwijl ze orders van grootte sneller draaien. Dit maakt het realistisch om enorme bibliotheken van kandidaat-kleurstoffen, OLED-emittenten en fluorescente sondes virtueel te screenen, wat de ontdekking van moleculen met precies afgestelde kleuren en helderheid versnelt.
Bronvermelding: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Trefwoorden: moleculaire optica, graf-neurale netwerken, machine-learning chemie, fluorescente kleurstoffen, oplosmiddeleleffecten