Clear Sky Science · en
A conformational benchmark for optical property prediction with solvent-aware graph neural networks
Why predicting molecule colors matters
From the bright pixels in phone screens to the dyes in solar cells and the glowing probes used to see inside living tissue, many modern technologies rely on molecules that absorb and emit light at just the right colors. Designing these molecules is hard: small changes in structure or solvent can shift their colors dramatically, and traditional quantum-chemistry calculations are too slow to guide large-scale searches. This paper introduces a new dataset and machine-learning models that treat molecules in full three dimensions and explicitly account for their surrounding liquid environment, enabling much faster and more accurate prediction of optical properties.
Building a better map of colorful molecules
The authors first assembled and cleaned a large collection of experimental data on how organic "chromophores"—the light-absorbing parts of molecules—behave in different solvents. They combined several public datasets and then painstakingly corrected problems such as invalid structural descriptions, inconsistent charge states, and misleading metal-containing structures. The result is nablaColors, covering 13,731 unique molecules and 26,369 chromophore–solvent pairs with measured absorption, and for many of them also emission wavelengths and light-emission efficiency (photoluminescence quantum yield). This careful curation reduces noise that can confuse machine-learning models and sets a reliable foundation for further study.
Adding the missing third dimension
Most existing machine-learning tools for predicting optical properties represent molecules as flat graphs: atoms are nodes and chemical bonds are lines. However, excited states and light absorption depend sensitively on real three-dimensional shapes—bond angles, twists, and weak interactions—which these 2D pictures cannot fully capture. To remedy this, the team generated 3D structures for every chromophore using a multi-step pipeline: an initial rough 3D layout, a faster semi-empirical quantum method, and then more accurate density-functional theory (DFT) optimizations, both in vacuum and with an implicit model of the surrounding solvent. This new 3D extension, nablaColors-3D, provides multiple conformations per molecule, each reflecting a different level of physical realism and computational cost. 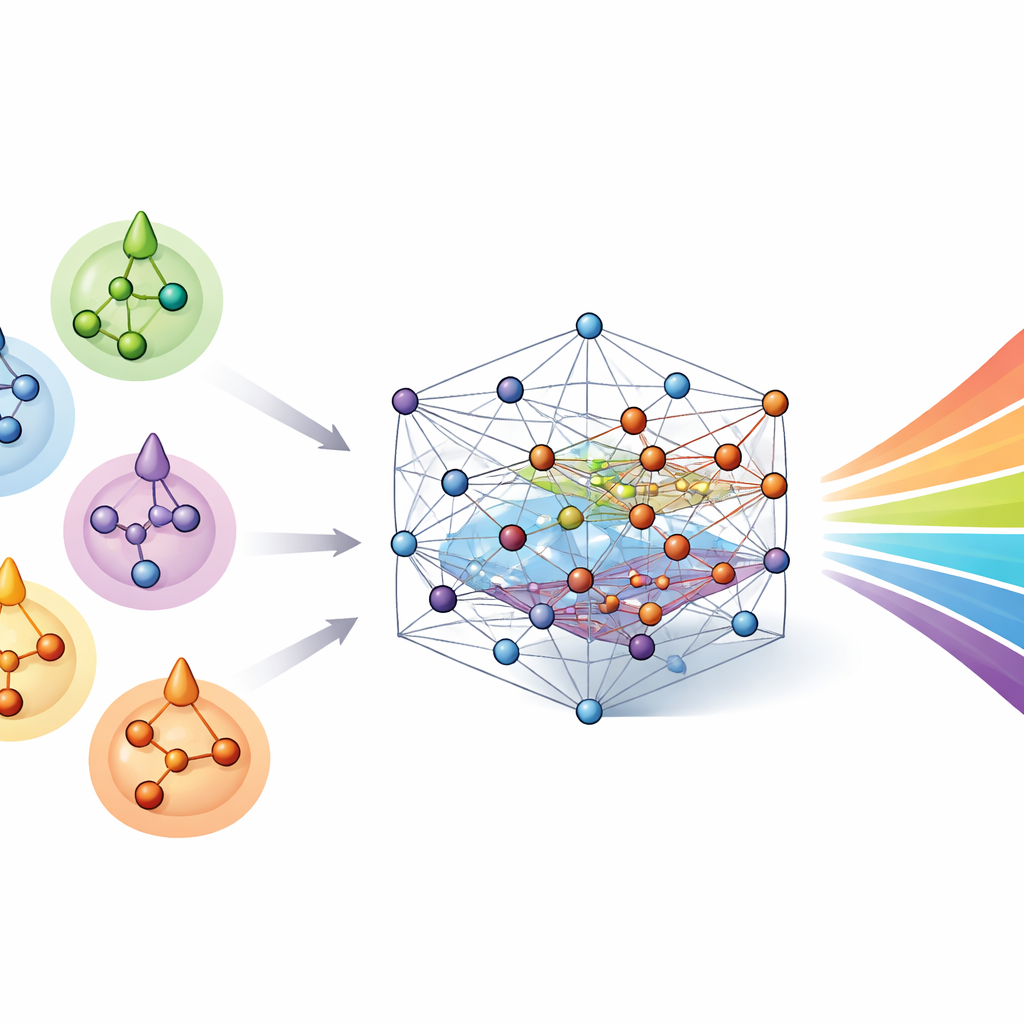
Teaching neural networks to see shape and solvent
With nablaColors-3D in hand, the authors built a benchmark to compare a range of machine-learning models, from established 2D graph neural networks to state-of-the-art 3D architectures that respect physical symmetries in space. They also designed a "solvent-aware" upgrade: a separate, lightweight neural network encodes the solvent’s structure from its own molecular representation, producing a compact solvent fingerprint. This fingerprint is combined with the chromophore’s 3D representation so that the main model can learn how the liquid environment subtly shifts the molecule’s geometry and electronic structure. By using a rigorous scaffold-based data split, the benchmark ensures that closely related molecules never appear in both training and test sets, so measured performance reflects true generalization rather than memorization.
How much geometry detail is enough?
An important practical question is whether it is worth paying the high computational price of very accurate geometries. The team systematically varied the type of 3D conformations given to each model—ranging from cheaper semi-empirical structures to more demanding DFT optimizations in vacuum and in implicit solvent—while keeping all training settings fixed. In general, better geometries tended to improve predictions, but the effect depended on the model and on whether explicit solvent fingerprints were used. Once solvent embeddings were included, performance differences between geometry sources shrank, showing that much of the solvent’s influence could be captured by this separate encoding rather than by ever more expensive conformer calculations. For their best model, they even showed that inexpensive structures generated by standard chemical software could replace quantum-optimized ones during training with almost no loss in accuracy. 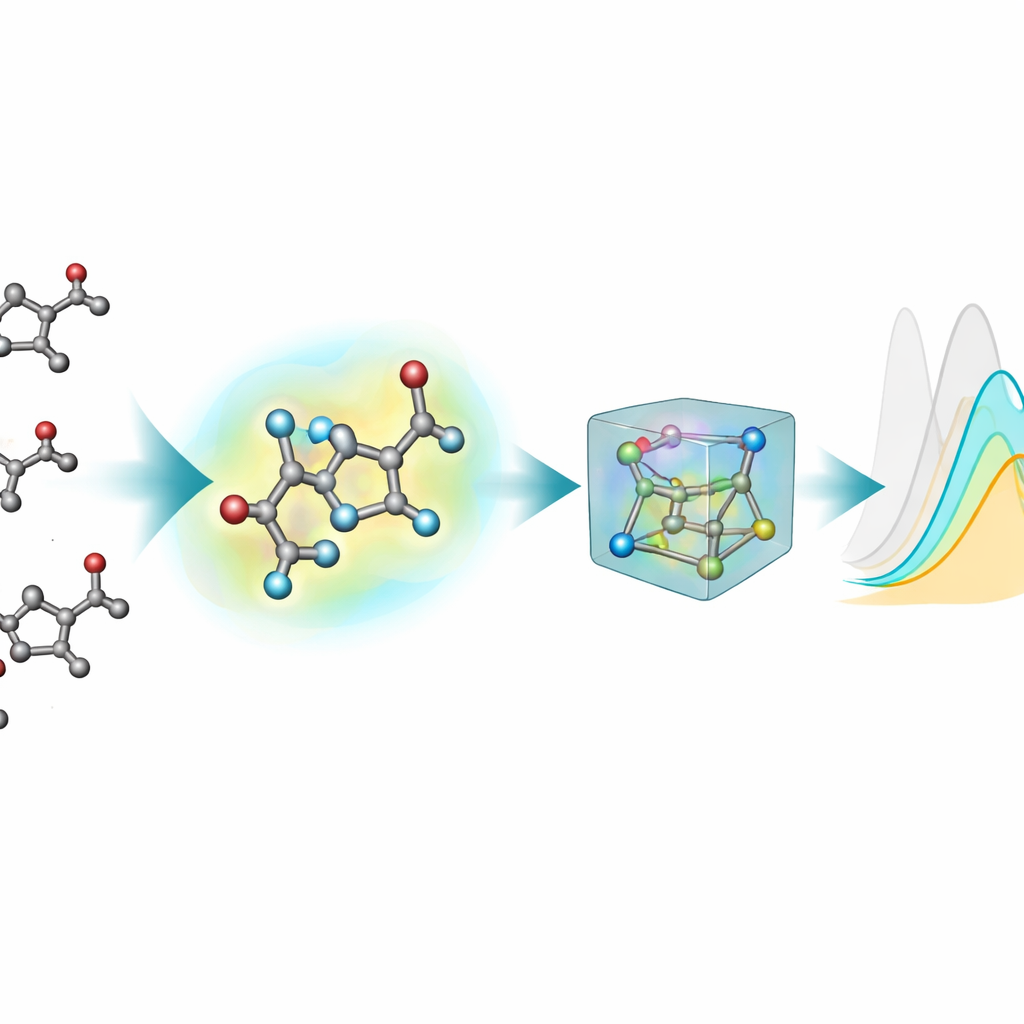
A leap beyond traditional methods
Among all tested models, a 3D transformer-based architecture called UniMol+—augmented with solvent embeddings into a variant the authors call UniProp—performed best. UniProp achieved a mean absolute error of about 16 nanometers for absorption wavelengths on a held-out test set, more than a 30% improvement over the strongest 2D baseline and far better than a widely used time-dependent DFT method, which erred by about 62 nanometers. Crucially, UniMol+ had been pretrained on large quantum-chemistry datasets to learn how to refine rough 3D structures toward high-level geometries. This "geometry-denoising" ability lets it accept relatively cheap conformers at prediction time while still capturing the fine structural details that matter for optical behavior.
Toward a universal optical design tool
Finally, the authors extended UniProp to predict not only absorption peaks, but also emission wavelengths and light-emission efficiency in a single multitarget model. It maintained high accuracy across all three properties, with only a slight trade-off for absorption, demonstrating that the same 3D features capture shared physical factors behind different photophysical processes. For non-specialists, the key takeaway is that three-dimensional, solvent-aware neural networks—trained on a carefully curated benchmark—can now outperform traditional quantum methods while running orders of magnitude faster. This makes it realistic to virtually screen huge libraries of candidate dyes, OLED emitters, and fluorescent probes, accelerating the discovery of molecules with precisely tuned colors and brightness.
Citation: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Keywords: molecular optics, graph neural networks, machine learning chemistry, fluorescent dyes, solvent effects