Clear Sky Science · pl
Benchmark konformacyjny do przewidywania właściwości optycznych z uwzględnieniem rozpuszczalnika przy użyciu grafowych sieci neuronowych
Dlaczego przewidywanie kolorów molekuł ma znaczenie
Od jaskrawych pikseli w ekranach telefonów po barwniki w ogniwach słonecznych i świecące sondy używane do obserwacji wnętrza żywych tkanek — wiele współczesnych technologii opiera się na molekułach, które absorbują i emitują światło o odpowiednich barwach. Projektowanie takich molekuł jest trudne: drobne zmiany w strukturze lub w rozpuszczalniku mogą dramatycznie przesunąć ich kolory, a tradycyjne obliczenia chemii kwantowej są zbyt wolne, by kierować szeroko zakrojonymi poszukiwaniami. Artykuł przedstawia nowy zbiór danych i modele uczenia maszynowego, które traktują molekuły w pełnych trzech wymiarach i explicite uwzględniają otaczające je środowisko ciekłe, umożliwiając znacznie szybsze i dokładniejsze przewidywanie właściwości optycznych.
Budowanie lepszej mapy barwnych molekuł
Autorzy najpierw zebrali i oczyścili dużą kolekcję danych eksperymentalnych dotyczących zachowania organicznych „chromoforów” — części molekuł absorbujących światło — w różnych rozpuszczalnikach. Połączyli kilka publicznych zestawów danych i skrupulatnie skorygowali problemy takie jak nieprawidłowe opisy struktur, niespójne stany ładunku oraz mylące struktury zawierające metale. Wynikiem jest nablaColors, obejmujący 13 731 unikalnych molekuł i 26 369 par chromofor–rozpuszczalnik z zmierzonymi długościami absorpcji, a dla wielu z nich także długościami emisji i efektywnością emisji światła (kwantowy zysk fotoluminescencji). Ta staranna kuracja redukuje szumy mogące mylić modele uczenia maszynowego i tworzy rzetelne podstawy do dalszych badań.
Dodanie brakującego trzeciego wymiaru
Większość istniejących narzędzi ML do przewidywania właściwości optycznych reprezentuje molekuły jako płaskie grafy: atomy jako węzły i wiązania chemiczne jako krawędzie. Jednak stany wzbudzone i absorpcja światła zależą wrażliwie od rzeczywistych trójwymiarowych kształtów — kątów wiązań, skrętów i słabych oddziaływań — których te dwuwymiarowe obrazy nie oddają w pełni. Aby to naprawić, zespół wygenerował struktury 3D dla każdego chromoforu przy użyciu wieloetapowego potoku: wstępnego, przybliżonego układu 3D, szybszej metody półempirycznej, a następnie dokładniejszych optymalizacji w teorii funkcjonału gęstości (DFT), zarówno w próżni, jak i z użyciem implicytnego modelu rozpuszczalnika. Nowe rozszerzenie 3D, nablaColors-3D, dostarcza wiele konformerów na molekułę, z których każdy odzwierciedla inny poziom realizmu fizycznego i kosztów obliczeniowych. 
Nauczanie sieci neuronowych rozpoznawania kształtu i rozpuszczalnika
Mając nablaColors-3D, autorzy zbudowali benchmark do porównania szeregu modeli ML — od ugruntowanych 2D grafowych sieci neuronowych po nowoczesne architektury 3D respektujące symetrie fizyczne w przestrzeni. Zaprojektowali też "świadome rozpuszczalnika" ulepszenie: oddzielna, lekka sieć neuronowa koduje strukturę rozpuszczalnika z jego własnej reprezentacji molekularnej, wytwarzając zwarte odcisk rozpuszczalnika. Ten odcisk łączy się z 3D reprezentacją chromoforu, tak by główny model mógł uczyć się, jak środowisko ciekłe subtelnie przesuwa geometrię i strukturę elektronową molekuły. Dzięki rygorystycznemu podziałowi danych opartemu na szkielecie (scaffold), benchmark zapewnia, że blisko spokrewnione molekuły nigdy nie pojawiają się jednocześnie w zestawach treningowych i testowych, więc mierzone wyniki odzwierciedlają prawdziwą generalizację, a nie zapamiętywanie.
Ile szczegółu geometrycznego wystarczy?
Istotne praktyczne pytanie brzmi, czy warto płacić wysoką cenę obliczeniową za bardzo dokładne geometrie. Zespół systematycznie zmieniał rodzaj konformerów 3D dostarczanych każdemu modelowi — od tańszych struktur półempirycznych po bardziej wymagające optymalizacje DFT w próżni i w implicytnym rozpuszczalniku — przy zachowaniu stałych wszystkich ustawień treningu. Generalnie lepsze geometrie miały tendencję do poprawiania przewidywań, ale efekt zależał od modelu i od tego, czy użyto explicite odcisków rozpuszczalnika. Po włączeniu osadzeń rozpuszczalnika różnice wydajności między źródłami geometrii zmniejszyły się, co pokazuje, że dużą część wpływu rozpuszczalnika można uchwycić przez tę oddzielną reprezentację zamiast przez coraz bardziej kosztowne obliczenia konformerów. W przypadku najlepszego modelu autorzy wykazali nawet, że niedrogie struktury generowane przez standardowe oprogramowanie chemiczne mogą zastąpić kwantowo zoptymalizowane podczas treningu niemal bez utraty dokładności. 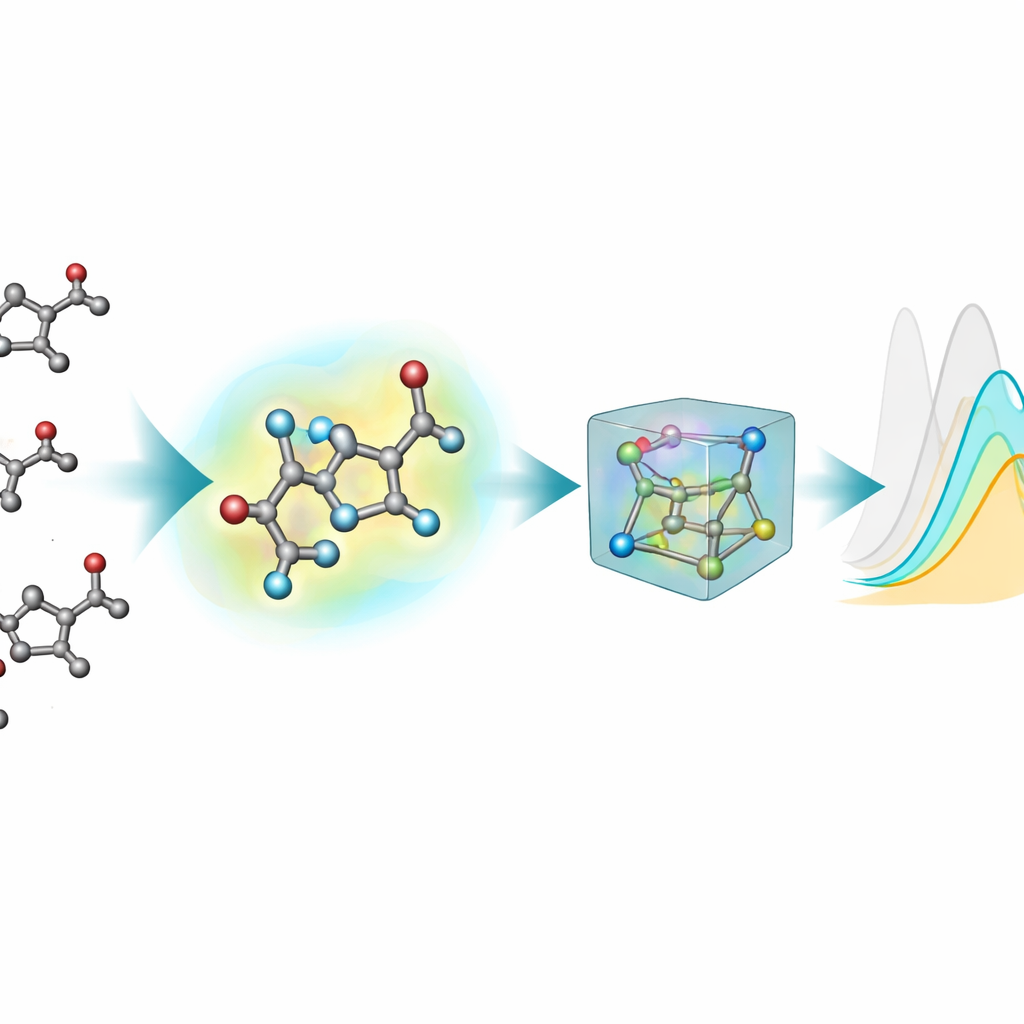
Krok dalej niż tradycyjne metody
Spośród wszystkich testowanych modeli najlepiej wypadła oparta na transformatorze architektura 3D nazwana UniMol+ — rozszerzona o osadzenia rozpuszczalnika w wariancie, który autorzy nazywają UniProp. UniProp osiągnął średni błąd bezwzględny około 16 nanometrów dla długości absorpcji na zestawie testowym, co stanowi ponad 30% poprawę względem najsilniejszej 2D bazy i znaczną przewagę nad powszechnie stosowaną metodą TD-DFT, która błędnie przewidywała o około 62 nanometry. Kluczowe jest to, że UniMol+ był wstępnie trenowany na dużych zbiorach danych chemii kwantowej, aby nauczyć się, jak przekształcać surowe struktury 3D w kierunku geometrii wysokiego poziomu. Ta zdolność "odszumiająca geometrię" pozwala mu przyjmować relatywnie tanie konformery w czasie predykcji, jednocześnie uchwytując drobne szczegóły strukturalne istotne dla zachowań optycznych.
W kierunku uniwersalnego narzędzia do projektowania optycznego
Na koniec autorzy rozszerzyli UniProp, aby przewidywał nie tylko piki absorpcji, lecz także długości emisji i efektywność emisji światła w jednym modelu multitargetowym. Model utrzymał wysoką dokładność dla wszystkich trzech własności, z jedynie niewielkim kompromisem dla absorpcji, co pokazuje, że te same cechy 3D chwytają wspólne czynniki fizyczne stojące za różnymi procesami fotofizycznymi. Dla niespecjalistów kluczowy wniosek jest taki, że trójwymiarowe, świadome rozpuszczalnika sieci neuronowe — wytrenowane na starannie skuratorskim benchmarku — mogą teraz przewyższać tradycyjne metody kwantowe, działając przy tym rzędy wielkości szybciej. To sprawia, że realistyczne staje się wirtualne przesiewanie ogromnych bibliotek kandydatów na barwniki, emitery OLED i sondy fluorescencyjne, przyspieszając odkrycie molekuł o precyzyjnie dostrojonych kolorach i jasności.
Cytowanie: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Słowa kluczowe: optyka molekularna, grafowe sieci neuronowe, chemia uczenia maszynowego, barwniki fluorescencyjne, efekty rozpuszczalnika