Clear Sky Science · es
Un banco de pruebas conformacional para la predicción de propiedades ópticas con redes neuronales gráficas conscientes del disolvente
Por qué importa predecir los colores de las moléculas
Desde los píxeles brillantes de las pantallas de los teléfonos hasta los tintes en celdas solares y las sondas luminosas usadas para observar tejidos vivos, muchas tecnologías modernas dependen de moléculas que absorben y emiten luz en colores muy concretos. Diseñar estas moléculas es difícil: pequeños cambios en la estructura o en el disolvente pueden desplazar mucho sus colores, y los cálculos clásicos de química cuántica son demasiado lentos para guiar búsquedas a gran escala. Este artículo presenta un nuevo conjunto de datos y modelos de aprendizaje automático que tratan las moléculas en tres dimensiones completas y tienen en cuenta explícitamente su entorno líquido, permitiendo predicciones de propiedades ópticas mucho más rápidas y precisas.
Construyendo un mapa mejor de moléculas coloridas
Los autores reunieron y limpiaron primero una gran colección de datos experimentales sobre cómo se comportan los «cromóforos» orgánicos —las partes de la molécula que absorben luz— en distintos disolventes. Combinón varios conjuntos de datos públicos y corrigió meticulosamente problemas como descripciones estructurales inválidas, estados de carga inconsistentes y estructuras engañosas que contenían metales. El resultado es nablaColors, que cubre 13.731 moléculas únicas y 26.369 pares cromóforo–disolvente con absorción medida, y para muchos de ellos también longitudes de emisión y eficiencia de emisión de luz (rendimiento cuántico de fotoluminiscencia). Esta curación cuidadosa reduce el ruido que puede confundir a los modelos de aprendizaje automático y establece una base fiable para estudios posteriores.
Añadiendo la tercera dimensión que faltaba
La mayoría de las herramientas de aprendizaje automático existentes para predecir propiedades ópticas representan las moléculas como grafos planos: átomos como nodos y enlaces químicos como líneas. Sin embargo, los estados excitados y la absorción de luz dependen de forma sensible de las formas tridimensionales reales —ángulos de enlace, torsiones e interacciones débiles— que estas imágenes 2D no capturan por completo. Para remediarlo, el equipo generó estructuras 3D para cada cromóforo mediante una canalización de varios pasos: un diseño 3D inicial aproximado, un método cuántico semiempírico más rápido y luego optimizaciones más precisas con teoría del funcional de la densidad (DFT), tanto en vacío como con un modelo implícito del disolvente circundante. Esta nueva extensión 3D, nablaColors-3D, proporciona múltiples conformaciones por molécula, cada una reflejando un distinto nivel de realismo físico y coste computacional. 
Enseñar a las redes neuronales a ver la forma y el disolvente
Con nablaColors-3D en mano, los autores construyeron un banco de pruebas para comparar una gama de modelos de aprendizaje automático, desde redes neuronales gráficas 2D consolidadas hasta arquitecturas 3D de vanguardia que respetan las simetrías físicas del espacio. También diseñaron una mejora «consciente del disolvente»: una red neuronal separada y ligera codifica la estructura del disolvente a partir de su propia representación molecular, produciendo una huella digital compacta del disolvente. Esta huella se combina con la representación 3D del cromóforo para que el modelo principal pueda aprender cómo el entorno líquido desplaza sutilmente la geometría y la estructura electrónica de la molécula. Usando una división de datos rigurosa basada en andamios (scaffolds), el banco de pruebas garantiza que moléculas estrechamente relacionadas nunca aparezcan en los conjuntos de entrenamiento y prueba al mismo tiempo, de modo que el rendimiento medido refleje verdadera generalización y no memorización.
¿Cuánto detalle geométrico es suficiente?
Una pregunta práctica importante es si merece pagar el alto coste computacional de geometrías muy precisas. El equipo varió sistemáticamente el tipo de conformaciones 3D dadas a cada modelo —desde estructuras semiempíricas más baratas hasta optimizaciones DFT más exigentes en vacío y en disolvente implícito— manteniendo fijos todos los ajustes de entrenamiento. En general, las geometrías mejores tendieron a mejorar las predicciones, pero el efecto dependía del modelo y de si se usaban huellas explícitas del disolvente. Una vez incluidas las incrustaciones (embeddings) del disolvente, las diferencias de rendimiento entre fuentes de geometría se redujeron, lo que muestra que gran parte de la influencia del disolvente puede capturarse con esta codificación separada en lugar de mediante cálculos de conformadores cada vez más costosos. Para su mejor modelo, incluso demostraron que estructuras económicas generadas por software químico estándar podían reemplazar a las optimizadas por métodos cuánticos durante el entrenamiento con casi ninguna pérdida de precisión. 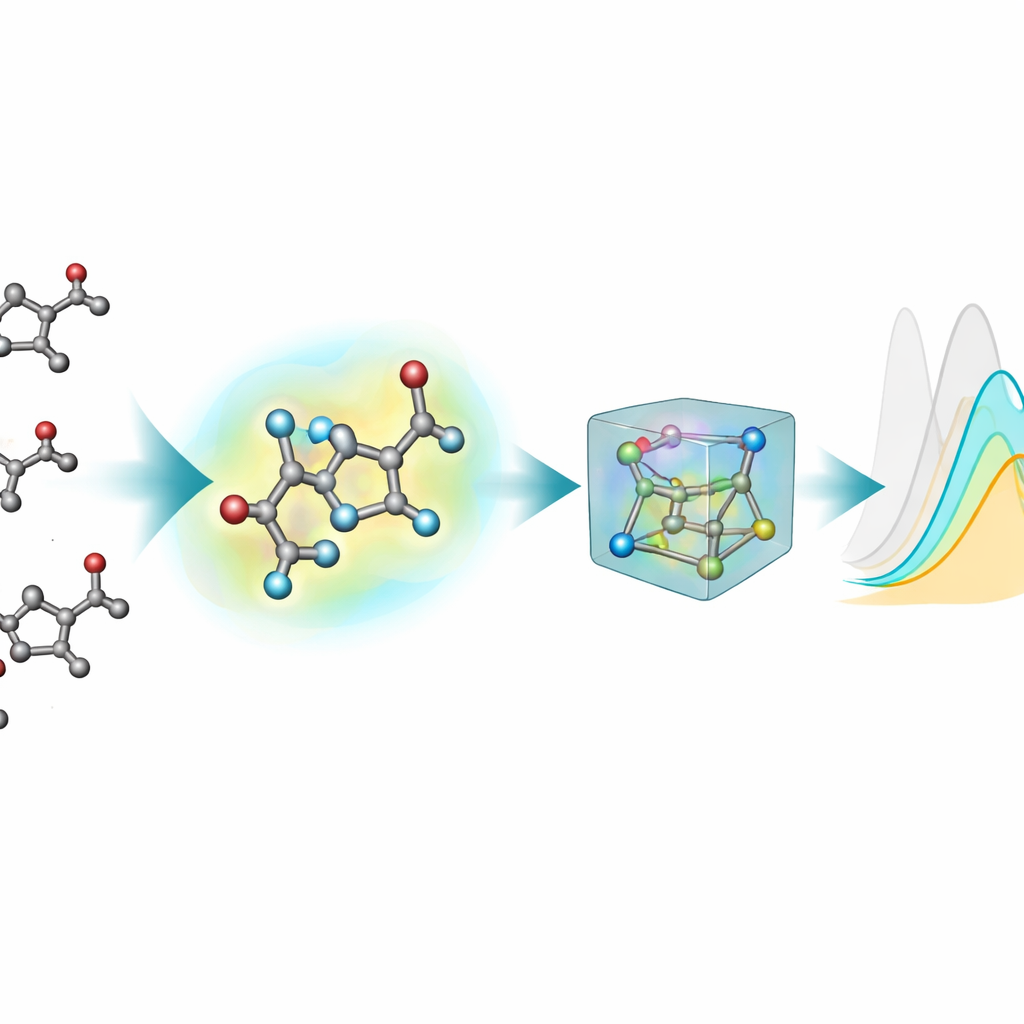
Un salto más allá de los métodos tradicionales
Entre todos los modelos probados, una arquitectura 3D basada en transformadores llamada UniMol+ —augmentada con incrustaciones de disolvente en una variante que los autores denominan UniProp— fue la que mejor rindió. UniProp alcanzó un error absoluto medio de alrededor de 16 nanómetros para longitudes de absorción en un conjunto de prueba retenido, más de un 30% de mejora frente a la mejor línea base 2D y muy por delante de un método ampliamente usado de DFT dependiente del tiempo, que falló por unos 62 nanómetros. Es crucial que UniMol+ había sido preentrenado en grandes conjuntos de datos de química cuántica para aprender a refinar estructuras 3D aproximadas hacia geometrías de alto nivel. Esta capacidad de «desruido geométrico» le permite aceptar conformadores relativamente baratos en tiempo de predicción mientras captura los detalles estructurales finos que importan para el comportamiento óptico.
Hacia una herramienta universal de diseño óptico
Finalmente, los autores ampliaron UniProp para predecir no solo picos de absorción, sino también longitudes de emisión y eficiencia de emisión de luz en un único modelo multitarea. Mantuvo una alta precisión en las tres propiedades, con solo una ligera compensación en absorción, demostrando que las mismas características 3D capturan factores físicos comunes detrás de distintos procesos fotofísicos. Para el público no especializado, la conclusión clave es que las redes neuronales tridimensionales y conscientes del disolvente —entrenadas en un banco de pruebas cuidadosamente curado— pueden ahora superar a los métodos cuánticos tradicionales mientras funcionan órdenes de magnitud más rápido. Esto hace realista el cribado virtual de enormes bibliotecas de tintes candidatos, emisores para OLED y sondas fluorescentes, acelerando el descubrimiento de moléculas con colores y brillo afinados con precisión.
Cita: Potapov, D., Rogovoi, S., Khrabrov, K. et al. A conformational benchmark for optical property prediction with solvent-aware graph neural networks. Commun Chem 9, 136 (2026). https://doi.org/10.1038/s42004-026-01944-5
Palabras clave: óptica molecular, redes neuronales gráficas, química con aprendizaje automático, tintes fluorescentes, efectos del disolvente